This is public documentation for the Materials Project (MP). The Materials Project is a decade-long effort from the Department of Energy to pre-compute properties of "materials" and make this data publicly available, with the intent of accelerating the process of materials discovery. In this context, a material can mean either an inorganic crystal (like silicon), or a molecule (like ethylene carbonate). Possible applications are vast, but might include better batteries, solar energy, water splitting, optoelectronics, catalysts and more (see here for a list of publications).
Table of Contents
Methodology
This section contains information on how we generate and validate our computed data sets.
Apps
This section talks about how we present information on the Materials Project website as "apps", and what these apps contain.
Getting Involved
The Materials Project is a public, collaborative project, offered free of charge. It only succeeds thanks to the efforts of everyone who participates! Visit this section to learn how to get involved.
Errors
If you notice an error or omission, please let us know at or directly via .
The Materials Project documentation is a living document and always a work in progress.
Glossary of Terms
Terms used by the Materials Project (MP), ordered alphabetically. Some terms are scientific terms while other terms refer to tools used in MP infrastructure.
Builder. A builder is a little script written in the Python programming language that helps create new database collection(s) from input database collection(s). It's typically used to allow common analysis tasks to be repeated automatically, for example the calculation of "energies above hull" when new calculations are added to the database. Builders are an essential step in the Materials Project database release process and are formalized with the emmet code.
Chemical system. On Materials Project, a chemical system is a set of materials whose members all contain the same elements. It is usually noted with as dash-delimited list of elements. For example, the "Ga-In-N" chemical system would contain all materials containing Ga, In or N or combinations of these elements (Ga, In, N2, GaN, InGaN, etc.).
Correction scheme. The Materials Project performs calculations using a simulation technique with known systematic errors. A correction scheme is employed to adjust energies based on the elements present in a material to address these systematic errors. Only elements for which sufficient experimental data is available can be corrected.
Energy above hull. A measure of a material's thermodynamic stability. This value refers to a mathematical construction that can be calculated from a set of formation energies and compositions known as a convex hull, and often referred to here as a "phase diagram." However, unlike most phase diagrams, convex hulls are usually given without a temperature axis since the simulation technique used (DFT) gives predictions at zero temperature. A material which lies "on the convex hull" is predicted to be thermodynamically stable, while off the hull is predicted to be metastable or unstable. Values above 200 meV/atom are considered very large and suggest an unstable material that might not be synthesizable, however this ceiling differs significantly by chemistry. Energies above hull are given as a guide and subject to both limits of calculation precision (several meV) and also of calculation accuracy due to limitations of the simulation technique used, where errors can be significant in certain chemistries.
Mixing scheme. The Materials Project uses two slightly different simulation techniques depending on the elements present in a material. These are GGA (Generalized Gradient Approximation) and GGA+U, where the +U (Hubbard correction) is a correction applied to address systematic deficiencies in GGA when simulating elements with highly localized electrons such as d-orbitals or f-orbitals. Energies from these respective techniques are not directly comparable with each other, so a mixing scheme is employed such that elements can be compared. Details of the mixing scheme can be found in this paper.
You can login to the Materials Project either using an existing social identity provider (currently GitHub, Google, Facebook, Microsoft or Amazon) or via an email link.
Be aware, your Materials Project account is linked to both your email address and the method that you log in. If you log in via a different method, this will be registered as a new account.
Here are some issues people have encountered when trying to sign in the website, and their solutions:
I want to log in with my social identity provider (GitHub/Google/Facebook/Microsoft/Amazon), but I can’t.
Ensure that your password for your provider is correct (go to their site and log in there), ensure that you have a full name set on that account, and ensure that you allow Materials Project to see your basic profile info (name and email address).
You also may be behind a firewall that doesn’t allow GitHub/Google/Facebook/Microsoft/Amazon. In that case, use our email based option instead.
I appear to sign in OK (the popup goes away), but then I remain on the sign-in screen.
How do I cite Materials Project?
Citations are appropriate wherever Materials Project data, methods or output are used. See this page on the Materials Project website for more information:
There is a canonical Materials Project citation, and additional citations for specific properties or tools. See also the page for information on how to cite a specific database version.
Where do the material properties shown on Materials Project come from?
The Materials Project core data is all calculated in-house by the Materials Project team using a variety of simulation methods. To understand the quality of these predictions, it is crucial to read the peer-reviewed publications from the Materials Project where each property is benchmarked as much as possible against known experimental values: this will give an estimate of typical error and, importantly, any systematic error that may be present.
Why are the lattice parameters different to what I expect?
The same crystal structure can have multiple, equivalent sets of lattice parameters depending on what crystallographic "setting" is used.
Typically, there are two sets of lattice parameters reported. Lattice parameters can be defined for the primitivecell, which is a definition of the crystal with the fewest number of atoms and therefore convenient for simulations and other uses, and the conventional cell, which is typically easier to visualize and more like you will see in textbooks.
If the lattice parameters are very different to what you expect, check the setting first!
Some systematic errors are also present. These will typically be an over-estimation of 1–3% for most crystals. Layered crystals will also typically have significant error in the interlayer distances since van der Waals interactions are not well-described by the simulation methods (PBE) used by Materials Project. These systematic errors will be improved as Materials Project switches to user newer simulation methods (r2SCAN). See for more information.
Why is the band gap different to what I expect?
Electronic band gaps are difficult to calculate reliably from first principles, especially using methods that scale well to hundreds of thousands of materials. The method used by the Materials Project (PBE) systematically underestimates band gaps.
While it would be possible to provide higher quality calculations for a select number of materials, with more accurate band gaps, it is noted that for materials discovery purposes it is useful to have a dataset that has the same systematic error. See Electronic Structure for more information.
Why has a value changed on Materials Project?
The Materials Project presents the data it generates in two ways:
As individual calculations. These are always the same, and as far as possible Materials Project tries to ensure all historical calculations remain available. Typically, only advanced users will access information about individual calculations.
As aggregated information. This is information generated from a combination of individual calculations. This information is what is presented on the public "material details" pages, and is what most users will access. As new, improved calculations are performed, this aggregated information can change.
The Materials Project periodically updates this aggregated information in the form of new database releases. See for information on the latest database releases.
If performing scientific research with Materials Project data, make sure to cite the database version from which the data was retrieved. See for more information.
What is a "task_id" and what is a "material_id" and how do they differ?
Every database needs a unique key which can be used to distinguish one entry from another. In the Materials Project, each unique material is given a material_id (also referred to in various places as mp-id, mpid, MPID). This allows a specific polymorph of a given material to be referenced. For example, wurtzite GaN is assigned the material_id of , while zinc blende GaN is assigned a material_id of .
How does a "material_id" get assigned?
The Materials Project is a computational resource. All of the information on a given material details page is actually a combination of data generated from many individual calculations or "tasks". It is also important that these tasks also have unique identifiers.
When a task is added to the Materials Project database, it will get an identifier assigned with the format mp-[0-9] ("mp-" with numbers after it). These identifiers are assigned sequentially, so smaller numbers usually refer to older calculations. An identifier referring to an individual calculation task are known as a task_id.
When the Materials Project database is built, a unique material will then have a collection of multiple different task_ids associated with it. The numerically smallest task_id will then become the material_id. This ensures that, as new, additional calculations are associated with the same material, its material_id should not change.
In the past, I have seen material_ids that start with "mvc", what are these?
Some calculation tasks were associated with a search for multivalent cathode materials. These tasks were given the prefix mvc- instead of mp- and thus some materials also had the prefix mvc-. However, this caused confusion and this approach has been retired. Tasks with the prefix mvc- still exist since the task_id cannot change, but a material_id will now always start with an mp- prefix by convention provided that at least one task associated with that material has the mp- prefix.
Do material_ids ever change? Do task_ids ever change?
A task_id will never change. It will always refer to the same, individual calculation task.
A material_id might change in rare instances, such as the removal of the mvc- prefix, although this is avoided wherever possible.
If a material_iddoes change, we ensure a redirect on the website is always in place, and the new material_id can also be found programmatically with the API using the get_material_id_from_task_id() function. This way, any publications or research that reference an older material_id are still valid, and the relevant data can still be retrieved.
What does ______ mean?
Consult our glossary here:
If a term is used in Materials Project but is not listed, and we will add it.
GGA/GGA+U Calculations
Details on GGA and GGA+U calculations run by the Materials Project
Details of calculation parameters for the density functional theory (DFT) calculation results contained in the Materials Project (MP) database.
We use DFT as implemented in the Vienna Ab Initio Simulation Package (VASP) software [1] to evaluate the total energy of compounds. For the exchange-correlational functional, we employ a mix of Generalized Gradient Approximation (GGA) and GGA+U, or a mix of GGA, GGA+U, and r2SCAN. Both mixing schemes are described here. All calculations are performed at 0 K and 0 atm. All computations are performed with spin polarization on and with magnetic ions in a high-spin ferromagnetic initialization (the system can of course relax to a low spin state during the DFT relaxation). For a select number of materials, alternate spin states are searched for. Details on this can be found in the Magnetic Properties section.
Input structures are sourced from many different places, including the Inorganic Crystal Structure Database (ICSD). [2] We relax all cell and atomic positions in our calculation two times in consecutive runs. When multiple crystal structures are present for a single chemical composition, we attempt to evaluate all unique structures as determined by an affine mapping technique. [3]
More detailed information on the GGA/GGA+U and r2SCAN calculations run by the Materials Project can be found in the following two subsections:
[1]: Kresse, G. & Furthmuller, J., 1996. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 54, pp.11169-11186.
[2]: G. Bergerhoff, The inorganic crystal-structure data-base, Journal Of Chemical Information and Computer Sciences. 23 (1983) 66-69.
[3]: R. Hundt, J.C. Schön, M. Jansen, CMPZ - an algorithm for the efficient comparison of periodic structures, Journal Of Applied Crystallography. 39 (2006) 6-16.
Energy Corrections
How energy adjustments and corrections are calculated on the Materials Project (MP) website.
To better model energies across diverse chemical spaces, we apply several adjustments to the total calculated energy of each material. These adjustments fall into two different sets, each of which is described in a different subsection. One set, consisting of anion and GGA/GGA+U mixing scheme corrections, and another consisting of only GGA/GGA+U/r2SCAN mixing scheme corrections. The former is used in the in the current and legacy data, while the latter is only present in releases after the addition of r2SCAN calculations (post v2022.10.28). Both of are used to produce ComputedStructureEntry objects, and mixed phase diagrams.
How optical absorption spectra are calculated on the Materials Project (MP) website.
The optical absorption spectra is obtained by calculating the frequency-dependent dielectric tensors using VASP. It uses independent particle approximation and assumes only vertical interband transitions to obtain the imaginary part of the dielectric tensors. Via the Kramers-Kronig relations the relationship between the dispersions of the real and imaginary parts of the dielectric function can be established. With both the real and imaginary part of the frequency-dependent dielectric tensors, one can calculate the optical absorption coefficient at different photon energies. Our results are validated against the experimental database: https://refractiveindex.info
Obtaining the charge density shown on the Materials Project (MP) website.
Charge density data is obtained directly from the CHGCAR files that are output by our static DFT calculations. For more detailed information about this data see the VASP wiki.
An isosurface visualization of the charge density can be found on the material details pages. To obtain the full set of volumetric data for a given material the API should be used.
Thermodynamic Stability
Grain Boundaries
How grain boundaries are calculated on the Materials Project (MP) website.
To do.
DFT Parameters
Description of the density functional theory (DFT) parameters used in MOF calculation results displayed on the Materials Project (MP) website.
We use density functional theory (DFT) as implemented in the Vienna Ab Initio Simulation Package (VASP) 5.4.4. All calculations are carried out at 0 K and 0 atm. The plane-wave kinetic energy cutoff was set to 520 eV, which is 1.3 times the highest cutoff recommended among the PAW PBE pseudopotentials we use. Unless stated otherwise, we used a k-point mesh of 1000/(number of atoms per cell), computed and arranged using Pymatgen. The geometries were considered converged when the net forces were all less than 0.03 eV/Å. Gaussian smearing of the band occupancies as applied with a smearing width of 0.01 eV. Symmetry considerations were disabled. In general, a high-spin magnetic initialization was applied with 5 µB for d-block elements (excluding Zn, Cd, Hg), 7 µB for f-block elements (excluding Lu, Lr), and no magnetic character for the remaining elements. A local minimum magnetic configuration was found in each case, although there may be a lower energy global minimum for systems with complex magnetic orderings.
For additional calculation details, refer to the VASP files made available on NOMAD.
MOF Methodology
Overview of methodology for metal-organic framework (MOF)-related calculations and analyses on the Materials Project (MP).
Alloys
How alloy data is calculated on the Materials Project (MP) website.
Description of the density functional theory (DFT) functionals and level of theory used in MOF calculation results displayed on the Materials Project (MP) website.
In all cases, the geometries are DFT-optimized structures at the PBE-D3(BJ) level of theory, and all properties are derived from single-point (i.e. static) calculations on these PBE-D3(BJ) optimized structures. In general, most properties are presented at the PBE-D3(BJ) level of theory. However, certain properties (e.g. band gaps, partial charges) for select materials are also provided based on HLE17, HSE06*, and HSE06 single-point calculations on the PBE-D3(BJ) optimized structures. Conventionally, these would be referred to as PBE-D3(BJ), HLE17//PBE-D3(BJ), HSE06*//PBE-D3(BJ), and HSE06//PBE-D3(BJ), respectively. However, for brevity, we typically refer to them as PBE, HLE17, HSE06*, and HSE06. The PBE functional is a generalized gradient approximation (GGA) functional, HLE17 is a high-local-exchange meta-GGA functional, HSE06 is a screened hybrid functional with 25% Hartree-Fock (HF) exchange, and HSE06* is the same as HSE06 but with 10% HF exchange. For computational efficiency, the HLE17, HSE06*, and HSE06 calculations were carried out with a k-point grid of 500/(number of atoms per cell).
Synthesis Explorer
Search synthesis recipes extracted from literature sources by natural language processing.
This section presents some basic information about the Battery Explorer app on MP and a short tutorial of how to use it.
QMOF IDs
What is a QMOF ID?
Each material in the QMOF Database is assigned a unique 7-digit QMOF ID that is associated with that material. All calculations associated with a given QMOF ID are for a given PBE-D3(BJ) optimized structure. Each QMOF ID represents a unique structure, as determined using Pymatgen's StructureMatcher. As such, the primitive unit cells of any two structures are distinct after any relevant volume rescaling. Depending on your personal definition of a unique material, you may wish to further unique-ify the structures. For instance, MOFs with closed-pore and open-pore configurations would be considered unique, MOFs with different linker configurations would be considered unique, and so on.
Topology
What is a MOF topology, and how was it determined?
The MOF topology describes the number of vertices, edges, and connectivity of the MOF building blocks. Several thousand topologies can be explored on the Reticular Chemistry Structure Resource. In the QMOF Database, the topology is detected using MOFid based on the available topologies in the RCSR as of June 1st, 2019.
Property Definitions
Catalysis Explorer
Structure Details
Neat structures! Tell me a bit about them?
Materials Explorer
Calculation Parameters
What VASP settings were used?
Tutorials
Visual walkthroughs of common use cases of the Phase Diagram App on Materials Project.
Phase Diagram
Background, tutorials, and FAQ for the Phase Diagram App on the Materials Project (MP) website.
Background
It may take a few seconds, depending on your connection, to actually get logged in. This is because we have an external identity provider verify your email address so that we don’t have to store any passwords on our servers.
You may also have an older browser that won’t work well with our website at all. The latest version of Mozilla Firefox, Google Chrome, or Microsoft Edge will work well. Older versions of Internet Explorer will not work.
I tried using the email option several times but haven’t received a login link.
We currently don’t do any validation of your email addresses, so if it “looks right”, i.e. you mistype [email protected] as myname@gmali.com, we will still try to send to the wrong address. Also check your "Spam" or "Junk" folder in case the login email has been flagged.
There is a known issue with Tencent @qq.com addresses, where Tencent throttles delivery and you might not get an email within a reasonable amount of time. Please consider using an alternative to your @qq.com address for login.
Resources for getting assistance with your MP questions and needs.
Materials Science Community Forum at matsci.org
The Materials Project runs a forum at matsci.org intended as a shared space for several computational materials science projects, as well as general discussion about materials science. For the past several years, this effort has been co-run by the OpenKIM project. See About matsci.org for more information about the forum and its governance.
Enquiries via email are always welcome, but please consider using the first: if questions or feedback are asked in a public setting, it allows others to benefit from seeing the answer too, and allows more people to participate in the conversation.
Potential Collaborators
The Materials Project welcomes collaborations and strives to maintain an environment where people are encouraged to share their findings as well as their analysis methods.
If you are interested in collaborating with others or are seeking ways to actively contribute:
Join the weekly infrastructure update Zoom call and listen to decisions being made to improve the Materials Project or bring up a specific item to discuss. To request to attend, email us with the subject line "Request to Join MP Update Call" and a brief introduction as well as the specific item you would like to discuss. Depending on the topic proposed, it might be referred for discussion on the Materials Project forum instead.
Materials Project hosts annual meetings for discussions among Materials Project Principal Investigators, their research groups, and the infrastructure team. If you have a suggestion for an item to be discussed in this context, please also send us an email. If you are a new member of the Materials Project collaboration, reach out to us so that you can get involved in these meetings directly.
Reach out to in the Materials Project, especially if you are already contributing to code on (for example, ) and would like to get in work with people who maintain/review these repositories. You can read more about their involvement, field of expertise, current projects and see if their goals align with yours to propose areas of collaboration.
Code of Conduct
Guidance for conduct within the Materials Project (MP) organization.
The Materials Project does not have a unified code of conduct at present since it is a joint, collaborative effort, and different aspects of the Materials Project, such as its different open-source codes, are led and maintained by different individuals at different institutions.
However, as guidance, we refer all contributors to the Contributor Covenant for setting expectations for each other. Text from the Contributor Covenant is copied below.
We have set up the [email protected] email address for any issues involving inappropriate conduct.
Our Pledge
We as members, contributors, and leaders pledge to make participation in our community a harassment-free experience for everyone, regardless of age, body size, visible or invisible disability, ethnicity, sex characteristics, gender identity and expression, level of experience, education, socio-economic status, nationality, personal appearance, race, caste, color, religion, or sexual identity and orientation.
We pledge to act and interact in ways that contribute to an open, welcoming, diverse, inclusive, and healthy community.
Our Standards
Examples of behavior that contributes to a positive environment for our community include:
Demonstrating empathy and kindness toward other people
Being respectful of differing opinions, viewpoints, and experiences
Giving and gracefully accepting constructive feedback
Examples of unacceptable behavior include:
The use of sexualized language or imagery, and sexual attention or advances of any kind
Trolling, insulting or derogatory comments, and personal or political attacks
Public or private harassment
Explore and Search Apps
These apps are for exploring and searching the datasets available in Materials Project. This section provides an overview, tutorials, and FAQ for each of the Explore and Search appson the Materials Project (MP) website.
Most data in "Explorer" apps are generated directly by Materials Project, but some are contributed by third parties, such as the Catalysis Explorer by the Open Catalyst Project, and the MOF Explorer, by Andrew Rosen et al.
Documentation Credit
Acknowledgements for the individuals who helped write the Materials Project documentation.
The Materials Project documentation is a collaborative effort between Materials Project staff, contributors, and researchers including graduate students, postdocs and members of the Materials Project community.
A recent list of contributors can be found here:
See also the "Documentation Authors" sections on individual documentation pages.
Molecules Methodology
Overview of methodology for molecules-related calculations and analyses on the Materials Project (MP).
There is new molecules data planned for release on Materials Project. This documentation will include information on this once the data has been released.
For existing molecules data, please refer to the following publications:
Website Changelog
2022-12-16 (7ca3bcd3)
Fix issue with API query, see .
Parameters and Convergence
Parameter and convergence details for GGA and GGA+U calculations run by the Materials Project
Calculation Parameters
We use the Projector Augmented Wave (PAW) method for modeling core electrons with an energy cutoff of 520 eV. This cutoff corresponds to 1.3 times the highest cutoff recommended among all the pseudopotentials we use (more details can be found in the ). A baseline k-point mesh of 1000/(number of atoms in the cell) is used for all computations. Specifically, the Monkhorst-Pack method is used for the k-point choices (with -centered for hexagonal cells), and the tetrahedron method is used to perform the k-point integration. It is important to note that Pymatgen has the ability change those default parameters if they are not adequate for the computation (e.g., switch to another k-point integration scheme). Some details of our calculation method can be found in ref ; however, the Materials Project has updated many parameters as documented throughout the Methodology sections. The most up-to-date input sets can be
r2SCAN Calculations
Details on r2SCAN calculations run by the Materials Project
Since database release v2022.10.28 the Materials Project has incorporated metaGGA functionals into its core dataset in the form of r2SCAN calculations. Part of this includes a that allows for the mixing of GGA, GGA+U and r2SCAN results in its thermodynamic data.
All r2SCAN data is obtained from a two-step workflow which is comprised of an initial GGA structure optimization, followed by final optimization with r2SCAN. The first step allows for the generation of an initial guess of the structure and charge density at a lower computational cost, speeding up the subsequent metaGGA calculation. More specifically, PBESol is used as the GGA functional for the first optimization step. For more details on the workflow see Ref .
Information regarding calculation parameters, convergence, and pseudopotential choices can also be found in the following subsections:
X-ray Absorption Spectra (XAS)
How x-ray absorption spectra are calculated on the Materials Project (MP) website.
X-ray Absorption Spectra (XAS) is calculated using the code FEFF.Feff is an ab initio multiple-scattering code for calculating excitation spectra and electronic structure. It is based on a real space Green’s function approach including a screened core-hole, inelastic losses and self-energy shifts, and Debye-Waller factors. The spectra include extended x-ray absorption fine structure (EXAFS), x-ray absorption near edge structure (XANES), and then both are stiched together to give a total XAS spectra. In addition the code can treat relativistic electron energy loss spectroscopy (EELS).
Multiple parameters are checked for convergence, including:
Self-consistent field (SCF)
Magnetic Properties
How magnetic properties are calculated on the Materials Project (MP) website.
What are magnetic properties?
The magnetic behavior of a material is a complex and rich research area. The Materials Project currently only addresses a narrow aspect of the magnetism of materials: the magnitude and ordering of atomic magnetic moments in a crystal structure, at zero temperature.
At present, Materials Project only considerscollinear magnetic order which means that atomic magnetic moments are represented by scalar values and not vectors.
Getting Involved
How to contribute to the Materials Project.
The Materials Project would not be the resource it is today without the sustained efforts of many individual contributors who have helped make the Materials Project better. The Materials Project is a free, academic resource, with only a small team of core maintainers: any help received is always appreciated, and means we can make the Materials Project better for everyone!
There are several ways to get involved:
If you are a software developer, you can join us on GitHub at . Improvements ("pull requests") and bug reports are welcome.
DFT Workflow
How to run a density functional theory (DFT) workflow for calculating / optimizing MOFs.
If you wish to run a QMOF-compatible workflow, we currently recommend using , which has a QMOF "recipe" available at from quacc.recipes.vasp.qmof.
First, install QuAcc via pip install quacc[vasp]. The QMOF workflow can be run via the following code-block after the setup process is completed:
Background
Components
Search
The Battery Explorer app, just like the Materials Explorer app, provides a search bar where one can search by chemical formula (eg. "CoO2") or by chemical system (eg. "Fe-P-O"). The user can also click on the periodic table to add elements to the search.
Overview
An overview of materials methodology.
This section provides a list of methodologies used in computational materials science to calculate properties of materials.
What is a material?
The term materials is used quite loosely, and has become more inclusive as the materials science community branches out to do more research in various areas of physics and chemistry. The conventional textbook definition of materials is divided, by chemical composition, into three classes: metals, ceramics and polymers.
Metallic materials are composed of, as the name suggests, metals. This class of materials is commonly seen in applications where structural integrity is important; jet engines, for example, has to use an alloy of up to 15 types of metals to withstand the high temperature generated by the combustion while still being able to stay structurally intact.
Tutorial
Visit ;
Enter the search criteria in the search box (labeled in red), or select elements from the periodic table of elements:
3. Click "Search" button to show search results.
Background
The materials synthesis recipes came from scientific literature through text mining and natural language processing approaches[1].
The synthesis recipes can be searched by the target material formula, precursor material formula, keywords (eg. ball-milled, impurities) and synthesis procedures (eg. synthesis type, performed operations, heating temperature etc.). Each entry gives the information about the target and precursors materials, the reaction equation, the synthesis procedure and the link to the source publication.
References
Structural Fidelity
Some nuances about structures in the QMOF Database (and all MOF databases, in fact)
As described in the original , the structural fidelity of MOF crystal structures is an incredibly challenging but important factor to consider when constructing DFT-based property databases. Many experimental MOF crystal structures have missing atoms (e.g. missing H atoms), under or overbonded atoms, unresolved disorder, charge-imbalances (e.g. missing or too many ions), and related issues. Similarly, some hypothetical MOF databases have building blocks with under or overbonded carbon atoms due to faulty functionalization routines. While significant effort was put into maximizing the structural fidelity of materials on the QMOF Database, we acknowledge that there are inevitably structures in the database that are not pristine.
If you find a material with poor structural fidelity, we ask you to listing the QMOF IDs of any problematic structures along with an explanation of the structural error. While we are not in a position to correct structures at this time, they will be removed from the QMOF Database when identified by the community, and a new version of the database will be minted.
Finite Temperature Estimation
Description of the methodology used to estimate Gibbs free energies of formation at finite (T>0 K) temperature. This is an option available within the Phase Diagram App.
Background
Methodology
Pore Geometry
How were pore-based properties computed?
Pore-related properties were computed using 0.3 with the high-accuracy flag (except in the rare cases where this failed, in which case the standard accuracy was used). These properties were computed using the PBE-D3(BJ) optimized structure.
The pore-limiting diameter is the smallest spherical diameter of void space that a guest species would need to traverse in order to diffuse through the material, whereas the largest cavity diameter is the largest spherical diameter that can fit within the void space of the material.
MOF Explorer
Predicted properties for metal–organic frameworks (MOFs) and coordination polymers, derived from the QMOF Database.
Background
MOFs are highly tunable materials composed of inorganic ions or clusters ("nodes") connected by organic ligands ("linkers") that yield a crystalline structure. To date, tens of thousands of MOFs have been experimentally synthesized, and virtually unlimited more can be hypothesized based on plausible node and linker building blocks.
The MOF Explorer () provides an interactive interface to the Quantum MOF (MOF) Database, which contains DFT-computed properties for ~20,000 MOFs and related MOF-like materials.
Electronic Structure
How were electronic structure properties computed?
Band gaps are computed using Pymatgen's , which uses the Kohn-Sham eigenvalues to compute the energy gap. In all cases, the displayed band gap is from a self-consistent calculation. We note that band gaps using the PBE functional are typically underpredicted compared to experiment. Although available for only a portion of the QMOF Database, band gaps calculated with the HSE06 functional are likely to be more accurate.
Density of states: TBD.
FAQ
Known issue: there is a problem generating Pourbaix diagrams for Zn-S . This is under investigation.
Symmetry
How is the symmetry determined?
The symmetry of each MOF is determined using Pymatgen's with a symprec tolerance of 0.1. The symmetry is based on the PBE-D3(BJ) optimized structure. It should be noted that all structure relaxations were carried out without explicit symmetry constraints of any kind.
Crystal Toolkit
Background, tutorials, and FAQ for the Crystal Toolkit app on the Materials Project (MP) website.
"Crystal Toolkit" refers to two things:
An app on the Materials Project that allows manipulation and transformation of crystal structures, both from the Materials Project database or uploaded by the user.
It refers to the , which was developed to write this app and which now powers the entire Materials Project website!
FAQ
A repository for frequently asked questions (FAQs) and their answers for the Phase Diagram App.
How do I visualize phase diagrams with 5 or more elements?
Analysis Apps
This section provides an overview, tutorials, and FAQ for each of the on the Materials Project (MP) website.
Pourbaix Diagram
Background, tutorials, and FAQ for the Pourbaix Diagram App on the Materials Project (MP) website.
In this section, we review how to use the Pourbaix Diagram app to generate Pourbaix diagrams for different elements. The app can generate diagrams for up to 4 elements at a time. We show generating the Fe and Fe-Cr Pourbaix diagram and illustrate some of the Advanced Options available in the app.
Here are the articles in this section:
Accepting responsibility and apologizing to those affected by our mistakes, and learning from the experience
Focusing on what is best not just for us as individuals, but for the overall community
Publishing others’ private information, such as a physical or email address, without their explicit permission
Other conduct which could reasonably be considered inappropriate in a professional setting
Ceramic materials are mostly oxides of metals, for the purpose of materials science. Some staple ceramic materials include Lead Zirconate Titanate (PZT) and CoO2. The former is the most commonly used piezoelectric (this type of materials converts mechanical work into electrical work) while the latter is the most commonly used Lithium ion battery cathode.
Polymer materials are results of polymerization of organic monomer molecules. As a relatively new member of the materials class, polymers have received much attention in the research space thanks to their versatility. Everyday plastic items, ranging from plastic bags, take out containers and Tupperware to water bottles, toys and Legos, are all polymers. Furthermore, polymer research in materials science also branch out to biological areas like drug delivery and tissue regeneration.
Another way to classify materials is by its usage case; in this scenario materials are classified into structural and functional materials. Structural materials, as the name suggests, serves to protect the structural integrity of something. A car frame, for example, would be a structural material. Functional materials, on the other hand, serves some kind of function (other than supporting weight, that is). The majority of modern-day materials science research lives in this functional materials space, ranging from semiconductors in computer chips, battery electrodes and OLEDs to piezoelectrics and MRI machines.
In short, materials science focuses on the joint of physics and chemistry and works on coming up with designs that satisfy a particular need in our real world.
What kind of properties do we care about?
Depending on the intended usage of our calculated data, there are different sets of properties that we care about.
For example, a materials scientist might be working on coming up with a semiconducting material that serves a certain purpose. They will be interested in looking at the electronic structure behavior of materials, such as band structure. Someone else might be interested in looking at piezoelectric properties, while others are interested in the migration behavior of a battery material. In short, depending on the interest, there is a range of properties we care about and calculate.
How do we calculate/predict these properties?
In computational materials science, we use Ab Initio (from first principle) methods to simulate the behavior of particles in the systems we're interested in. For materials data on the Materials Project, the majority of our work is done using Vienna Ab Initio Simulation Package (VASP), which implements Density Functional Theory (DFT) to calculate all kinds of properties from first principle.
Citations
References
How can I download a picture of the phase diagram?
Full multiple scattering (FMS)
EXCHANGE: The EXCHANGE card specifies the exchange correlation potential model used for XANES calculation.
COREHOLE: The COREHOLE card is used for specifying how the core is treated during XANES calculation.
Parameter-free calculations of x-ray spectra with FEFF9, J.J. Rehr, J.J. Kas, F.D. Vila, M.P. Prange, K. Jorissen, Phys. Chem. Chem. Phys., 12, 5503-5513 (2010)
Ab initio theory and calculations of X-ray spectra, J.J. Rehr, J.J. Kas, M.P. Prange, A.P. Sorini, Y. Takimoto, F.D. Vila, Comptes Rendus Physique 10 (6) 548-559 (2009)
Theoretical Approaches to X-ray Absorption Fine Structure, J. J. Rehr and R. C. Albers, Rev. Mod. Phys. 72, 621, (2000)
If you are a domain expert, you can join the discussion and help answer questions of less experienced users in our forum at https://matsci.org/materials-project.
If you are a domain expert, you can also notify us of errors, either in our public forum or via email at [email protected]. Please check our FAQ first to ensure that this error is not already known; some common issues arise from a misunderstanding of the data that Materials Project offers.
If you generate data, either experimental or computational, you can use our contribution platform MPContribs to upload and link your data to the relevant material on Materials Project. This helps us by being able to offer a more complete and helpful resource, and also helps improve the discoverability of your own research by making it available to a wider audience. All uploaded data is credited to the original authors, and will have links to the appropriate publications.
If you are an advanced user of Materials Project data or codes, you can help us improve documentation and tutorials.
If you have discovered or know about a new crystal structure that is not present in the Materials Project database, you can submit it to us for calculation to help us offer a more complete database. If you are an advanced user, we may be able to receive calculations directly, but this typically requires prior communication and planning.
Any help is gratefully received, and we work hard to try to give back to the community ourselves wherever possible!
Frequently, one is interested in identifying a MOF with a specific common name (e.g. HKUST-1, MOF-5, MOF-74). While common names cannot directly be queried in the MOF Explorer, MOFid/MOFkey can be used to carry out such a query using the following general procedure:
Download the CIF of the desired MOF from the published literature (e.g. from the original source publication). Some common MOFs can be found here.
Calculate the MOF's unique MOFid or MOFkey using the web-based ID Tool by simply uploading the structure and clicking submit. Please read the tips on the MOFid webpage carefully.
Copy down the MOFid and/or MOFkey information.
Query the MOF Explorer by SMILES (i.e. MOFid) or MOFkey. If there are multiple options, take the one you like. If multiple entries are returned in the MOF Explorer with the same reduced chemical formula, we generally recommend the structure with the lowest energy (per atom). This would represent the lowest energy conformer at the PBE-D3(BJ) level of theory.
There are other, slightly less comprehensive, ways of searching for a given MOF. For instance, you can search by DOI on the MOF Explorer, so if you know the DOI of the paper that reported the crystal structure of your MOF of interest, you can query by that. Additionally, if you know the CSD Refcode for a given MOF, you can query by that as well.
.
Total energy convergence
As mentioned, we currently employ a k-point mesh of 1000 per reciprocal atom (pra). However, we have performed a convergence test of total energy with respect to k-point density and convergence energy difference for a subset of chemically diverse compounds for a previous parameter set, which employed a smaller k-point mesh of 500 pra. Using a 500 pra k-point mesh, the numerical convergence for most compounds tested was within 5 meV/atom, and 96% of compounds tested were converged to within 15 meV/atom. Results for the new parameter set will be better due to the denser k-point mesh employed. Convergence will depend on chemical system; for example, oxides were generally converged to less than 1 meV/atom. [2]
Structure convergence
The energy difference for ionic convergence is set to 0.0005 * natoms in the cell. Data on expected accuracy on cell volumes can be found in a previous paper. [1] We have found these parameters to yield well-converged structures in most instances; however, if the structures are to be used for further calculations that require strictly converged atomic positions and cell parameters (e.g. elastic constants, phonon modes, etc.), we recommend that users re-optimize the structures with tighter cutoffs or in force convergence mode.
Authors
Shyue Ping Ong
References
[1]: A. Jain, G. Hautier, C. Moore, S.P. Ong, C.C. Fischer, T. Mueller, K.A. Persson, G. Ceder., A High-Throughput Infrastructure for Density Functional Theory Calculations, Computational Materials Science. vol. 50 (2011) 2295-2310.
[2]: L. Wang, T. Maxisch, G. Ceder, Oxidation energies of transition metal oxides within the GGA+U framework, Physical Review B. 73 (2006) 1-6.
import covalent as ct
from ase.io import read
from quacc.recipes.vasp.qmof import qmof_relax_job
# Read a MOF CIF
mof = read("mymof.cif")
# Make a QMOF-compatible job with on-the-fly error handling
workflow = ct.lattice(qmof_relax_job)
# Dispatch the workflow to the Covalent server
# with the Atoms object as the input
dispatch_id = ct.dispatch(workflow)(mof)
# Fetch the result from the server, if present
result = ct.get_result(dispatch_id)
print(result)
The Materials Project approaches magnetism in two ways:
Historically, all materials are initialized in a ferromagnetic configuration by default. This was a pragmatic choice due to the computational expensive of considering all possible magnetic ordering. During the simulation of these materials, it is possible that the magnetic order will converge to a non-ferromagnetic order, but more likely the order will remain ferromagnetic even if the true ground state of the material is non-ferromagnetic. Therefore, the reported magnetic order for most materials on the Materials Project is a description of the calculated magnetic order, and not a prediction of the true ground state magnetic order.
For some materials, Materials Project has started to systematically search for ground state magnetic ordering of materials. This means that multiple magnetic ground states are considered for each material: ferromagnetic, anti-ferromagnetic, ferrimagnetic, etc. So far, this systematic search has been done for several thousand magnetic oxides. For these materials, the reported magnetic order is therefore a prediction of the true ground state magnetic order.
References
Advanced Options
On the left tab on the app, users can choose to filter query results by composition and working ion, as well as battery properties such as average voltage or capacity.
Visualization Viewer
The battery material details page provides a visualization for the host material of the battery.
Data Table
The search result data table provides info on each entry, including formula, volume change, capacity and energy, etc.
Thermodynamic data
4. The molecular information is shown within each entry, by clicking on the Molecule ID:
Molecule information
5. Users can refine the search result via Filter, located on the top left part of the search results page. The filter can be applied to either the composition, or the basic properties.
Kononova, Olga, Haoyan Huo, Tanjin He, Ziqin Rong, Tiago Botari, Wenhao Sun, Vahe Tshitoyan, and Gerbrand Ceder. "Text-mined dataset of inorganic materials synthesis recipes." Scientific data 6, no. 1 (2019): 1-11.
Related links
Schematic representation of synthesis “recipes” extraction pipeline from reference 1
Diffraction Patterns
How diffraction patterns are calculated on the Materials Project (MP) website.
Introduction
Diffraction occurs when waves (electrons, x-rays, neutrons) scattering from obstructions act as secondary sources of propagation. In the case of crystal structures, atoms in periodic lattices act as scattering sites from which constructive and destructive interference can occur. Line spectra of scattering intensity as a function incident angle can give powerful information into the planar spacing and symmetries of a crystalline material.
X-ray Diffraction Formalism
The calculation of x-ray diffraction patterns (XRD) in the Materials Project relies on the diffraction condition in reciprocal space:
where is the wave vector of the incident x-ray, is the wavevector is the scattered x-ray and is the reciprocal lattice vector of the parallel set of diffracting planes with miller indices hkl. The length of reciprocal lattice plane vector is given by:
where is the wavelength of the x-ray. Therefore, the maximum diffraction plane condition which is searched is . Once all of the relevant diffraction planes with reciprocal lattice vectors within this limit are selected, the diffraction condition for each of these planes can be calculated:
where is the is the spacing of the hkl plane. The structure factor for each of these diffraction conditions is calculated as:
where is the index for the atoms in the unit cell, is the basis vector for the atoms in the unit cell. The atomic scattering factor is given by:
where , and are parameters fitted to individual elements. The intensity of each diffraction condition is given by the squared modulus of the structure factor.
Finally the Lorentz-polarization factor is applied to correct for the change in x-ray amplitude due to scattering angle and geometry of the experimental conditions:
Electron Diffraction
The Transmission Electron Microscopy (TEM) pattern for multiple Laue zones is calculated similarly to the XRD diffraction patterns and is available through the diffraction properties tab in the materials explorer.
References
[1]: De Graef, Marc, and Michael E. McHenry. Structure of materials: an introduction to crystallography, diffraction and symmetry. Cambridge University Press, 2012.
Community Resources
Links to Materials Project (MP) Community Resources and information about the MP Workshop.
The Materials Project organizes workshops where you can register to learn about data, various tools, and code infrastructure used to power the Materials Project. MP Workshops are designed for all user levels, including scientists with limited prior coding experience. The workshop is held over two days to provide an interactive experience where attendees are encouraged to follow along live coding demonstrations with workshop helpers available for individualized support and troubleshooting. There is also an optional one-day primer for those new to python programming or using MongoDB databases.
These workshops are normally held once a year in August with registration opening in the spring. The latest announcements regarding the MP Workshop is posted to the . Unfortunately there will not be a MP Workshop in 2022 due to the Materials Project Triennial Review.
Past MP Workshop Materials
To increase open access for the scientific the community, recordings and materials from the Materials Project Workshops are released publicly.
GGA/GGA+U/r2SCAN Mixing
Details on the GGA/GGA+U/r2SCAN mixing scheme corrections
An updated energy correction scheme [1] is used to allow for the mixing of GGA, GGA+U, and r2SCAN calculations. This is constructed by considering all electronic energies to be the sum of a reference energy, and a relative energy. The reference energy (Eref) for each functional is defined as the (empirically corrected) electronic energy of the GGA(+U) ground-state structure at each point in composition space. The energy of a material associated with either functional can then be expressed as a difference relative to a specific reference energy (ΔEref). The formation energy of a material is calculated in the usual way by subtracting the electronic energies of the elemental endpoints in each respective functional. It should be noted that ΔEref is calculated from the differences in polymorph energies, and consequently does not depend on the elemental endpoint energies. While the updated mixing scheme is similar to the previous scheme involving only GGA and GGA+U calculations, it extends the approach to be amenable to any two functionals without relying on pre-fitted energy correction parameters.
Mixing Rules
The two rules used to construct mixed GGA/GGA+U/r2SCAN phase diagrams are as follows:
Start with a GGA(+U) convex energy hull. Replace GGA(+U) energies with r2SCAN energies by adding their to the corresponding GGA(+U) reference energy.
Construct the convex energy hull using formation energy calculated using r2SCAN energies, only when r2SCAN calculations exist for every reference structure (i.e. every stable GGA(+U) structure). In this case, add any missing GGA(+U) materials by adding their to the corresponding r2SCAN reference energy.
For more detailed information on the mixing scheme and its benchmarks, see the original publication in Ref .
References
[1] Kingsbury, R.S., Rosen, A.S., Gupta, A.S. et al. A flexible and scalable scheme for mixing computed formation energies from different levels of theory. npj Comput Mater 8, 195 (2022).
Changes and Updates
A changelog of Materials Project (MP) updates to the website, documentation, database, and API.
The Materials Project is an active, academic research project. Changes are common as new research methods become available, and the quality and kind of data we present changes, and also as a result of organizational needs. This page summarizes major changes in different aspects of the Materials Project.
Upcoming Changes
This documentation will continue to be improved. New documentation is currently being written for each of the Materials Project "apps". Some pages may be blank until this is completed.
Previous Changes
Database
The Materials Project database is constantly evolving as new and better calculations become available, both as a result of new features and better methods, and also as errors or problems are identified and fixed.
See the following documentation page for a list of changes to the Materials Project database:
API
The Materials Project API has recently undergone a significant modernization effort. The new Materials Project website is exclusively powered by this API.
See the following documentation page for more information:
Website
The Materials Project has recently undergone a major change in its website architecture. More information on this can be seen in the .
It is recommended that the URL is used as the primary location of the Materials Project website, however a specific website version can be visited via the following links:
will always take visitors to the latest Materials Project website with the newest database version available.
will take visitors to a frozen snapshot of the older Materials Project website. This is powered by an older version of the database with known issues. The legacy website is being left online for some time as we fully transition to the next-gen website, and to allow users time to make any adjustments as necessary for features that may only be available on the legacy website, however the legacy website will be taken offline in due course.
See the website changelog for a detailed list of recent changes:
Documentation
The Materials Project documentation has gone through several iterations, powered previously by MediaWiki and MkDocs software. The current version is powered by GitBook. This switch was made to allow more easy and rapid changes to the documentation, in the hopes of ensuring documentation is maintained at a consistent, high quality.
The current documentation is also available via GitHub at . Edits and improvements from external users are very welcome, please submit a "pull request" with any suggest change or use the "Edit in GitHub" button on the relevant page.
The previous MkDocs documentation is for the historical record, and the older MediaWiki documentation are currently offline but available on request. However, the current version of the documentation should contain all necessary information including historical information. An effort has been made to ensure URLs remain the same during the transition from the previous MkDocs-powered documentation to the new GitBook-powered documentation.
Chemical Potential Diagrams (CPDs)
Overview of how chemical potential diagrams (CPDs) are constructed and visualized. These are available as part of the Phase Diagram App.
Introduction
The chemical potential diagram is the mathematical dual to the compositional phase diagram. To create the diagram, convex minimization is performed in energy (E) vs. chemical potential (μ) space by taking the lower convex envelope of hyperplanes. Accordingly, “points” on the compositional phase diagram become N-dimensional convex polytopes (domains) in chemical potential space.
For more information on this specific implementation of the algorithm, please cite/reference the paper below:
Todd, P. K., McDermott, M. J., Rom, C. L., Corrao, A. A., Denney, J. J., Dwaraknath, S. S., Khalifah, P. G., Persson, K. A., & Neilson, J. R. (2021). Selectivity in Yttrium Manganese Oxide Synthesis via Local Chemical Potentials in Hyperdimensional Phase Space. Journal of the American Chemical Society, 143(37), 15185-15194.
References
[1] Yokokawa, H. “Generalized chemical potential diagram and its applications to chemical reactions at interfaces between dissimilar materials.” JPE 20, 258 (1999).
[1] Todd, P. K., McDermott, M. J., Rom, C. L., Corrao, A. A., Denney, J. J., Dwaraknath, S. S., Khalifah, P. G., Persson, K. A., & Neilson, J. R. (2021). Selectivity in Yttrium Manganese Oxide Synthesis via Local Chemical Potentials in Hyperdimensional Phase Space. Journal of the American Chemical Society, 143(37), 15185-15194.
SMILES, MOFid, and MOFkey
What are SMILES strings, a MOFid, and a MOFkey?
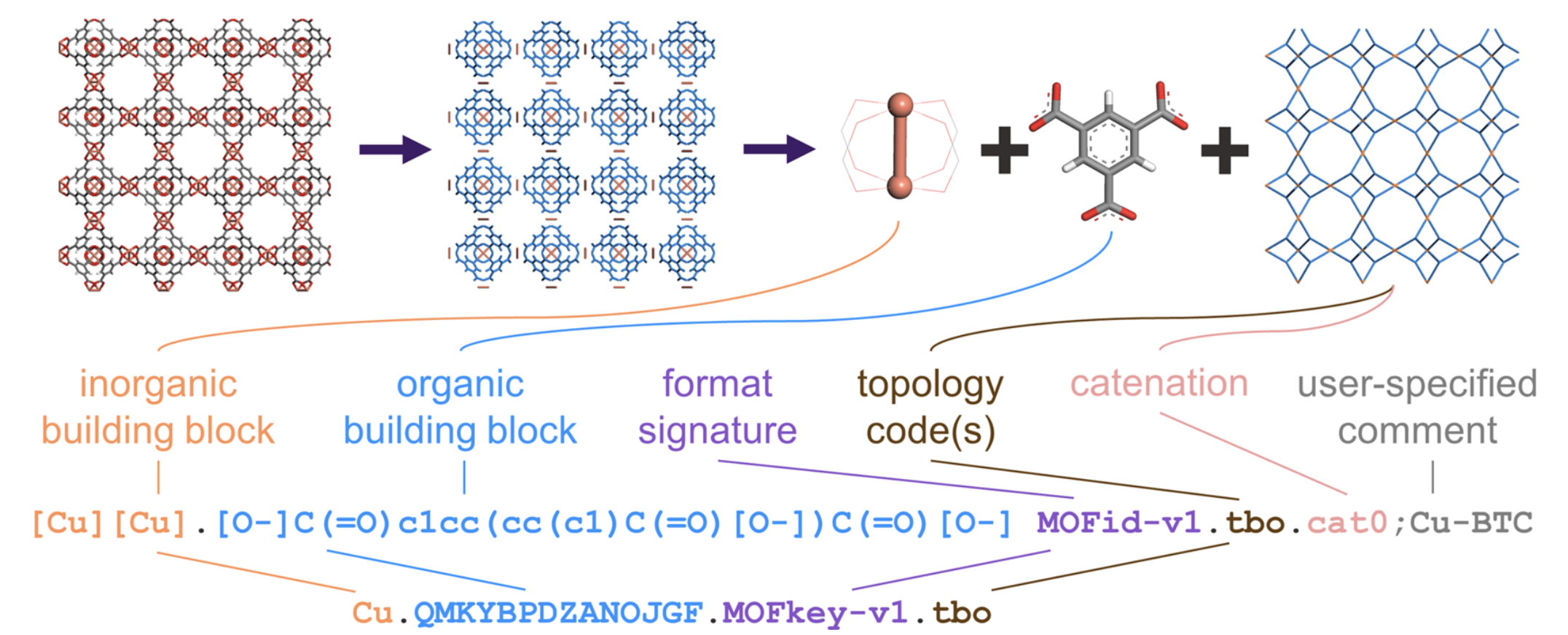
In prior work by Bucior et al., a pair of methods known as MOFid and MOFkey are described that can be used to assign a unique name for a given MOF. MOFid works by deconstructing a MOF into its node(s), linker(s), and topology. The nodes and linkers are represented as SMILES strings, the topology is determined using Systre, and any catenation is noted. These factors are combined into a single unique "MOFid". The MOFkey is simply a shorter, InChI-based hash of the MOFid. These methods are shown below for HKUST-1 (also known as Cu3(btc)2 and Cu-BTC):
Example MOFid and MOFkey for the MOF with common name HKUST-1.
The MOFid code is available here with a web-based version available here.
The SMILES search on the MOF Explorer is a partial-match of the MOFid. As such, one can query by just the node, just the linker, or even a substructure of the linker. If the user wishes to supply both a node and linker query, they should be provided in the MOFid format (separated by a "." with the node(s) listed before the linker(s)).
Population Analyses and Bond Orders
How were partial atomic charges, atomic spin densities, and effective bond orders computed?
All partial atomic charges are computed at the PBE-D3(BJ) geometry using one of several population analysis methods: Bader, DDEC6, and CM5. The DDEC6 and CM5 charges were calculated from Chargemol 09-26-2017. In all cases, the partial atomic charges are calculated from the DFT-computed charge density. In the QMOF Database, partial atomic charges are calculated using a charge density at one of four several levels of theory: PBE, HLE17, HSE06*, and/or HSE06. In general, the different levels of theory predict similar partial atomic charges. The different charge partitioning schemes, however, can result in very different partial atomic charges.
We report multiple magnetic properties for each material, including a net magnetic moment, atomic magnetic moments from VASP, and atomic spin densities calculated using the Bader and DDEC6 methods. In general, a high-spin magnetic initialization was provided (similar to what is done for the Materials Project). We note, however, that this does not mean all materials have high-spin character, as the initial magnetic moments are adjusted until they converges to a local minimum energy configuration in VASP. For applications that are highly reliant on an accurate description of the magnetic character, we acknowledge that there may be a lower energy magnetic configuration not captured via this initialization procedure.
Bond orders were computed using the method. Bond orders displayed in the MOF Explorer are the effective bond orders for each atom (i.e. a single value representing the sum of bond orders between the atom of interest and its neighbors). For the full list of bond orders between every pair of atoms, we refer the user to the DDEC6 files made available on NOMAD. For the crystal structure visualization, bond orders are not taken into account; instead, the coordination environments are determined from Pymatgen's .
Tutorial
Tutorial on using Catalysis Explorer
In this section, we will review how to use the Catalysis Explorer app of the Materials Project. The Catalysis Explorer allows for visualising structures with surface adsorbates and provides adsorption energies for those structures.
To begin, click the above link to go to the Catalysis Explorer app.
2. Search by composition
One of the ways of searching for a particular surface is through the bulk formula, within the composition tab. For example, you could search for Ti2Pd3.
3. Search by Adsorbate
Choose a certain adsorbate based on the SMILES or IUPAC formula. For example, if you were interested in finding the adsorption energy for CH2, the adsorbate SMILES would be *CH2 and the IUPAC formula would be C1 H2.
5. Search by formula
In this tab, you can choose surfaces based on their formula, material ID corresponding to their bulk, miller indices of the surface (individually as h,k,l) and surface shifts.
6. Real example
Say we were interested in CH2* on Ti2Pd3. Input the options in points 2 and 3 of this tutorial to find the following search results from the database (note that the exact options might change in the future).
7. Visualize structure and adsorption energies
Background
Components
Search
You can search using the periodic table to select different elements you would like in your Pourbaix diagram. You can select up to 4 elements to build the Pourbaix diagram.
Advanced Options
Several advanced opttions are available. You can filter solids, display a heatmap, or change the composition.
Pourbaix Diagram Viewer
Citations
To cite the Pourbaix Diagram App on Materials Project, please include the main Materials Project citation, as well as the references below:
Patel, Anjli and Norskov, Jens and Persson, Kristin and Montoya, Joseph. Efficient Pourbaix diagrams of many-element compounds, Physical Chemistry Chemical Physics 45 (2019)
Singh, Arunima and Zhou, Lan and Shinde, Aniketa and Suram, Santosh and Montoya, Joseph and Winston, Donald and Gregoire, John and Persson, Kristin. Electrochemical Stability of Metastable Materials, Chemistry of Materials 29 (2017)
Persson, Kristin and Waldwick, Bryn and Lazic, Predrag and Ceder, Gerbrand. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states, Physical Review B 85 (2012)
How to Cite
Please appropriately cite our work if you find it useful!
How to Cite
If you use the MOF Explorer in your work, please cite the following two references:
A.S. Rosen, S.M. Iyer, D. Ray, Z. Yao, A. Aspuru-Guzik, L. Gagliardi, J.M. Notestein, R.Q. Snurr. "Machine Learning the Quantum-Chemical Properties of Metal–Organic Frameworks for Accelerated Materials Discovery", Matter, 4, 1578-1597 (2021). DOI: .
A.S. Rosen, V. Fung, P. Huck, C.T. O'Donnell, M.K. Horton, D.G. Truhlar, K.A. Persson, J.M. Notestein, R.Q. Snurr. "High-Throughput Predictions of Metal–Organic Framework Electronic Properties: Theoretical Challenges, Graph Neural Networks, and Data Exploration," npj Comput. Mat.,8, 112 (2022). DOI: .
Ref. 1 describes the original release of the Quantum MOF (QMOF) Database, which at the time of publication consisted of PBE-computed properties for ~14,000 MOFs. This is the primary reference for the QMOF Database.
Ref. 2 builds upon the QMOF Database by introducing the MOF Explorer application, raises the number of structures to ~20,000, introduces hypothetical MOFs to the QMOF Database, and supplements the PBE data with HLE17, HSE06*, and HSE06 static calculations for select materials.
In addition, if using the data in your own work, we recommend specifying the version number for reproducibility purposes. The current version number is included at the bottom of each MOF details page and corresponds to the current version of the QMOF Database on .
FAQ
What input file formats are supported?
Any input file format supported by pymatgen. This includes CIF, POSCAR, CSSR and pymatgen JSON. See here for more information. Additional file formats might be supported on request.
A transformation doesn't seem to be working?
This is a known issue in some instances, where transformations may time out for certain larger crystal structures or combinations of inputs. Any transformation taking longer than 30 seconds will time out.
Please ask on the if this is a problem for you. We are improving this functionality over time. For advanced users, all transformations are also available for use via .
Molecules Explorer
The Molecules Explorer app is a new feature of the Materials Project. It enables searching for molecules relevant for electrolyte applications. The API is similar to that of Materials Explorer, where the user can query by one of the following methods:
Elements only;
Chemical formula;
SMILES string;
Molecule ID.
Upon receiving the query, Molecules Explorer outputs the search result, including information of the relevant molecules based on the query. For each molecule, the following properties are provided:
Electron Affinity;
Ionization Energy;
SMILES string representation;
Tutorial
In this section, we present several ways to navigate through the Battery Explorer.
The Battery Explorer app allows users to filter candidate battery materials using chemical formula/composition, as well as properties such as maximum volume change, average voltage, capacity, stability etc.
In each individual page for a battery material, the user can find information regarding the material such as calculated properties, voltage curve, oxygen evolution graph and a visulization of the host material.
1.
Downloading the Data
How to download the data available on https://materialsproject.org/mofs
Downloading the QMOF Database
The recommended way of downloading much of the data underlying the QMOF Database is at the following Figshare repository: . The data on Figshare includes DFT-optimized geometries (in XYZ and CIF format) and several tabulated properties, such as energies, partial atomic charges (DDEC6, CM5, Bader), bond orders (DDEC6), atomic spin densities (DDEC6, Bader), magnetic moments, band gaps, and more. For reproducibility purposes, we recommend noting the version of the QMOF Database you have used. Note that a mirror of the QMOF Database made to be interoperable with the Materials Project is available on , which can be queried with the if desired.
Surface Energies
Introduction
Surface energy is a measure of the energy change associated with the breaking of intermolecular bonds in a bulk material to create a surface. In thermodynamically stable materials, the creation of a surface will always increase energy, otherwise there would be a thermodynamic driving force to create surfaces and the material would sublimate. In theory, surface energy is equal to half of the energy of cohesion (the energy needed to break all of the bonds required to form two new surfaces). However, this perfect cleaving of surfaces is rarely achieved. In reality, surfaces often rearrange and/or react with their surroundings to passivate or adsorb molecules or atoms to lower their surface energy from the theoretical cohesive energy value.
Figure 1. Rules for mixing GGA(+U) (blue) and r2SCAN (red) energies onto a single phase diagram. (left) r2SCAN energies are placed onto the GGA(+U) hull by referencing them to the r2SCAN energy of the GGA(+U) ground state via ΔEref. A, B, C, ad D represent different polymophs at a single composition, and polymorph A is the ground state. (right) r2SCAN formation energies are used to build the convex hull only when there are r2SCAN calculations for every GGA(+U) ground state.
Two dimensional (2-D) chemical potential diagram for the V-S chemical system. Energies are DFT-calculated energies directly acquired from MP database.
Three dimensional (3-D) chemical potential diagram for the V-S-O chemical system. Energies are DFT-calculated energies directly acquired from MP database.
Relationship between 3-D chemical potential diagram and predominance diagrams, which are 2-D views of the full three-dimensional chemical potential diagram surface. Figure by Matthew McDermott.
Formalism
Surface energy is calculated using a slab model where a supercell of a crystal is oriented such that a given facet of interest is created and then exposed to vacuum by removing atoms from the supercell. If we are interested in creating a surface with the plane (hkl) exposed, lattice vector transformations are performed on the supercell with lattice vectors a and b parallel to the exposed plane (hkl) and lattice vector c as close to perpendicular to the exposed plane as is feasbile. This new unit cell is referred to as the oriented unit cell. The atoms in the oriented unit cell must also be shifted in the c direction in order to expose all possible symmetrically distinct atomic terminations. This algorithm for generating slabs is implemented in pymatgen [1].
The surface energy γhklσ of facet (hkl) of a slab model is calculated as:
where Eslabhkl,σis the total energy of the slab with termination σ, Ebulkhklis the per atom total energy of the bulk oriented unit cell, nslab is the total number of atoms in the slab and A is the surface area of the slab. The bulk oriented unit cell's atomic positions as well as its volume are relaxed, whereas in the slab model, only the atomic positions are relaxed.
DFT Parameters
All DFT calculations are performed in the Vienna Ab-initio Simulation Package (VASP) with the projector augmented wave (PAW) method. Exchange correlation effects are modeled using the Perdew-Berke-Ernzerhof (PBE) generalized gradient approximation (GGA) funcitonal. All calculations are spin polarized using a plane wave cutoff energy of 400eV. Full details can be found in [2].
References
[1]: Ong, S. P. et al. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Computational Materials Science68, 314–319 (2013)
[2]: Tran, R., Xu, Z., Radhakrishnan, B. et al. Surface energies of elemental crystals. Sci Data3, 160080 (2016)
γhklσ=2⋅AslabEslabhkl,σ−Ebulkhkl⋅nslab
2. Search the chemical formula of interest
Step 2 screenshot
3. Choose the filter of choice under the "Composition" Tab on the left
Step 3 screenshot
4. Select the working ion of choice
Step 4 screenshot
5. One can delete filters by clicking "x" next to the filter
Step 5 screenshot
6. Try a different type of filtering requirement - chemical system
Step 6 screenshot
7. Under Battery Properties tab on the left, choose filter of choice
Step 7 screenshot
8. Filter by average voltage
Step 8 screenshot
9. Filter by stability of the discharged state
Step 9 screenshot
10. Change the x-axis for the voltage curve
Step 10 screenshot
11. Link to materials detail page for each voltage step
Additional files and properties beyond those hosted on Figshare (e.g. VASP inputs and outputs, density of states, charge densities) can be obtained from NOMAD and Globus, as described in more detail below.
Downloading the VASP Files
NOMAD
All VASP input and output files are made available on NOMAD at the following datasets:
Querying NOMAD by external_id will allow you to search by the unique QMOF ID available on the MOF Explorer. Including a supplemental query of datasets will allow the user to specify one of the four datasets listed above for a specified level of theory. Links to the NOMAD files for a given material are available on each material's detail page. See the "Calculation Parameters" section of the documentation for a description of the different levels of theory.
Please note that there may be more entries on NOMAD than in the MOF Explorer. This is because structures are occasionally removed from the QMOF Database if any structural fidelity issues are identified, but entries cannot be deleted from NOMAD.
To download an entire NOMAD dataset, switch from the default "Entries" view to "Datasets".
Globus - Charge Densities
Due to their large filesizes, charge densities are made available via a Globus endpoint. First, set up a collection end-point, which can include your local machine or a compute cluster with Globus installed. Then choose a path in the collection in which to store the files. Once this is set up, select the folders and/or files you wish to download from the QMOF collection and choose "Transfer or Sync to..." to have Globus transfer the files to your specified location.
2. Type the search criteria such as the composition, chemical formula or mp-id in the search box or click the elements from the periodic table below the search box
3. Use the filters in the left to filter the search results
4. Click on Search
5. Click on Columns to select what properties to show for the search results
6. Click on the mp-id to go to the material page
7. Use the right tool bar on the to change the visualization settings and export the structure file
8. To download the structure, click "Export as"
9. A summary of material properties is shown in the right
10. An auto-generated description of the material from generated using Robocrystallographer (https://github.com/hackingmaterials/robocrystallographer)
11. Click on Crystal Structure to look at the basic structure information
12. Click on Properties to look at the materials properties
See the Methodology section for how these properties were calculated
Background
Description of the components of the phase diagram app, the intended functionality of each component, and a short review of the origin of the phase diagram construction and MP thermodynamic data.
The Phase Diagram App allows a user to create and visualize compositional phase diagrams for 1, 2, 3, and 4 element chemical systems using Materials Project data. It is also possible to create and visualize the corresponding chemical potential diagrams for 2 and 3 element systems. The user has access to some customization features, such as 1) changing the style of plot, 2) selecting data calculated with a certain DFT functional, 3) and using machine learning (ML)-estimated finite temperature data.
Methodology
The methodology behind thermodynamic energy calculations, phase diagram construction, and chemical potential diagram construction has been extensively discussed in the Methodology section of the MP Docs. See the links below:
App Components
Search by chemical system
Phase diagrams are created by chemical system (i.e., a collection of elements). To create a phase diagram in the Phase Diagram App, first select a set of elements by typing them either as a single string separated by dashes, or by clicking the elements in the periodic table viewer (which will auto-populate the search box).
Warning
Phase diagrams can only be plotted for chemical systems containing 1-4 elements. It is still possible to create phase diagrams for 5 or more elements, but this feature is only currently available inpymatgen.
Visualization Viewer
Once a chemical system has been selected (e.g., Li-Fe-O), you will an illustration of the compositional phase diagram for your system of interest load in the the viewer. Within the viewer, you can switch to the chemical potential diagram to view the same phase equilibria but within chemical potential space (see for more information).
1) Phase Diagram
2) Chemical Potential Diagram
If you are
Configure Visualization
The phase diagram viewer can be configured
Advanced Options
Data Table
MPContribs
Introduction to MP's contribution platform MPContribs
MPContribs provides a platform and advanced programming interface (API) to contribute computational as well as experimental data to Materials Project. Data on MPContribs is collectively maintained as annotations to existing MP materials (or formulas and chemical systems), and automatically exposed to over 200,000 MP users. The platform serves as the backbone for data and apps contributed to MP while leaving full ownership and control over the data with contributors. Contributed data is automatically shown on MP's or its disambiguation pages for formulas and chemical systems. A dedicated landing page is provided for each MPContribs project which can be used to reference the dataset in journal publications through Digital Object Identifiers (DOIs) provided by MP in collaboration with the DOE Office of Scientific and Technological Information (). The MPContribs can be used to programmatically retrieve, upload and modify contributed data.
See below for a list of and an overview of its . Continue with the following sections in MP's documentation to learn more:
Suggested Substrates
How suggested substrates are calculated on the Materials Project (MP) website.
Introduction
Materials synthesis techniques such as Chemical Vapor Deposition, Molecular Beam Eptixay, Sputtering, etc. are prevalent in materials research. Synthesizing materials with these techniques comes with a challenge: how does one determine which subtrate to use?
Epitaxial growth of heterogeneous interfaces requires a fundamental understanding of the substrate material, film material, cleavage planes, lattice mismatches, and resultant stresses and strains. The Materials Project (MP) stores crystallographic information for each material in its database, calculated via First Principles Density Functional Theory. Each material's crystallographic information, in particular the surface termination lattice parameters, is especially useful to find the epitaxial matches between a desired material (film) and a corresponding substrate. The Suggested Substrates tool outputs the Miller Indices of the substrate and the film (target material) termination plane, the minimal co-incident area (MCIA), and Elastic Energy.
Phonon Dispersion
How phonon dispersion and phonon band structures are calculated/visualized on the Materials Project (MP) website.
Introduction
A phonon is a collective excitation of a set of atoms in condensed matter. These excitations can be decomposed into different modes, each being associated with an energy that corresponds to the frequency of the vibration. The different energies associated with each vibrational mode constitute the phonon vibrational spectra (or phonon band structure). The vibrational spectra of materials play an important role in physical phenomena such as thermal conductivity, superconductivity, ferroelectricity and carrier thermalization.
There are different methods to calculate the vibrational spectra from first-principles using the density functional theory formalism (DFT). It can be obtained from the Fourier transform of the trajectories of the atoms on a molecular dynamics run, from finite-differences of the total energy with respect to atomic displacements or directly from density functional perturbation theory (DFPT). The latter method is the one used in the calculations on the Materials project page.
Tutorial
1.
Tutorial
1.
Tutorial
Visit ;
On the crystal toolkit web page, one can query Materials Project by MP-ID, or upload a structure file. (of what format???)
3. The interactive graphic user interface allows the user to visualize and manipulate crystal structures.
In the density functional perturbation theory formalism the derivatives of the total energy with respect to a perturbation are directly obtained from the self-consistency loop [1] For a generic point q in the Brillouin zone the phonon frequencies ωq,m and eigenvectors Um(qκ′β) are obtained by solving of the generalized eigenvalue problem
where κ labels the atoms in the cell, α and β are cartesian coordinates and Cκα,κ′β(q) are the interatomic force constants in reciprocal space, which are related to the second derivatives of the energy with respect to atomic displacements. These values have been obtained by performing a Fourier interpolation of those calculated on a regular grid of q-points obtained with DFPT.
Thermodynamic properties
The vibrational density of states g(ω) is obtained from an integration over the full Brillouin zone
where n is the number of atoms per unit cell and N is the number of unit cells. The expressions for the Helmholtz free energy ΔF, the phonon contribution to the internal energy ΔEph, the constant-volume specific heat Cv and the entropy S can be obtained in the harmonic approximation [2]
where kB is the Boltzmann constant and ωL is the largest phonon frequency.
Calculation details
All the DFT and DFPT calculations are performed with the ABINIT software package [3,4].
The PBEsol [5] semilocal generalized gradient approximation exchange-correlation functional (XC) is used for the calculations. This functional is proven to provide accurate phonon frequencies compared to experimental data [6]. The pseudopotentials are norm-conserving [7] and taken from the pseudopotentials table Pseudo-dojo version 0.3 [8].
The plane wave cutoff is chosen based on the hardest element for each compound, according to the values suggested in the Pseudo-dojo table. The Brillouin zone is sampled using equivalent k-point and q-point grids that respect the symmetries of the crystal with a density of approximately 1500 points per reciprocal atom and the q-point grid is always Γ-centered [^9].
All the structures are relaxed with strict convergence criteria, i.e. until all the forces on the atoms are below 10−6 Ha/Bohr and the stresses are below 10−4 Ha/Bohr3.
The primitive cells and the band structures are defined according to the conventions of Setyawan and Curtarolo [10].
Citation
Guido Petretto, Shyam Dwaraknath, Henrique P. C. Miranda, Donald Winston, Matteo Giantomassi, Michiel J. van Setten, Xavier Gonze, Kristin A. Persson, Geoffroy Hautier, Gian-Marco Rignanese, High-throughput density functional perturbation theory phonons for inorganic materials, Scientific Data, 5, 180065 (2018). doi:10.1038/sdata.2018.65
References
[1]: Gonze, X. & Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density functional perturbation theory. Phys. Rev. B 55, 10355–10368 (1997)
[2]: C. Lee & X. Gonze, Ab initio calculation of the thermodynamic properties and atomic temperature factors of SiO2 α-quartz and stishovite. Phys. Rev. B 51, 8610 (1995)
[3]: Gonze, X. et al. First-principles computation of material properties: the Abinit software project. Computational Materials Science 25, 478 – 492 (2002)
[4]: Gonze, X. et al. ABINIT: First-principles approach to material and nanosystem properties. Computer Physics Communications 180, 2582 – 2615 (2009)
[5]: Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008)
[6]: He, L. et al. Accuracy of generalized gradient approximation functionals for density-functional perturbation theory calculations. Phys. Rev. B 89, 064305 (2014)
[7]: Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013)
[8]: van Setten, M., Giantomassi, M., Bousquet, E., Verstraete, M.J., Hamann, D.R., Gonze, X. & Rignanese, G.-M., et al. The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table (2018). Computer Physics Communications 226, 39.
[9]: Petretto, G., Gonze, X., Hautier, G. & Rignanese, G.-M. Convergence and pitfalls of density functional perturbation theory phonons calculations from a high-throughput perspective. Computational Materials Science 144, 331 – 337 (2018)
[10]: Setyawan, W. & Curtarolo, S. High-throughput electronic band structure calculations: Challenges and tools. Computational Materials Science 49, 299 – 312 (2010)
The table below lists the various MPContribs portals currently available. When you explore data or consider contributing your data to MP, please pick the portal that best aligns with it.
Portal
URLs
Description
MPContribs
Datasets that don't fall under the purview of the other portals
MPContribsML
Benchmark datasets for Machine Learning
Concepts
Each MPContribs deployment is organized into projects. The MP account creating the project becomes its owner. An owner can ask for the MP accounts of their collaborators to be given access to their project. A collaborator assumes the same level of permissions within a project as the owner.
A project contains a list of contributions to existing MP materials (or alternatively to formulas and chemical systems). It's in the owner's purview to decide what exactly constitutes a project. Often this will simply be an umbrella for a dataset containing contributions to MP materials that are comparable in their scientific context and thus are consistent in their data schema.
Any MP account can create (or be an owner of) a maximum of 3 projects at any time. Project owners can immediately start adding up to 500 contributions to their project without approval from MP. To add more contributions, project owners or their collaborators can reach out to MPContribs administrators to obtain approval.
By default, projects are set to private, i.e. only visible to owners and their collaborators. Each individual contribution in a project is set to public by default and thus automatically released to the public when the project is published. Since the public/private flag can be controlled for each contribution individually, some contributions in a project can be kept private even if the project is public. The public/private state of a project and its contributions can be changed/reverted at any time.
A single contribution constitutes a small blob of data assigned and linked to the according MP material through identifiers such as MP's materials IDs, formulas or chemical systems. In addition to these identifiers, each individual contribution can contain the following four components:
A data component containing hierarchically organized key-value data (think nested dictionaries). In its flattened format, this component can contain a maximum of 50 keys/fields each of which becomes a column in the overview table on the project landing pages. Nested fields in the data dictionary are organized as nested columns on the landing page table. Any data types included in the data component become queryable, filterable and sortable using a wide variety of operators. Also see the API documentation for any MPContribs deployment for a generic list of available filters.
A structures component containing a list of up to 10 pymatgen structures with optionally customized names. A string in the format used for Crystallographic Information Files (CIFs) is stored with each structure and can be retrieved through the API or downloaded through the project landing pages.
A tables component containing a list of up to 10 . This component is intended for the inclusion of 2D spectra (think CSV files) with each contribution. A is generated for each table and included in the according contribution detail page for visualization purposes. Each DataFrame's name and other attributes (title, axis labels, ...) needed to configure the Plotly graph can be controlled via the Dataframes' attrs attribute. The total number of table rows is stored and all table cells formatted automatically. The API paginates the table rows for more efficient data retrieval. Each table can be downloaded as CSV programmatically or through the project landing pages.
An attachments component containing a list of up to 10 with customized names. Attachments can be gzipped text files (CSV, JSON, ...) or images in PNG, JPEG, GIF, or TIFF formats. An attachment can either be created directly from a file path or from a python list or dictionary using the mpcontribs.client.Attachment.from_data() method. Each attachment can be up to 2.4 MB large. Attachment meta-data are queryable but not its contents (think e-mail attachments).
Duplicate structures, tables, and attachments are only saved once internally but referenced by all contributions they were submitted with. See the section about contributing data for more information and examples.
Suggested Substrates tool in MP relies mainly on the geometrical principles of lattice matching, based off of Zurr and McGill [1].
Suppose there is two slabs of materials: a film and a substrate. The MP database the lattice parameters for both the film and substrate bulk crystal. Slabs are generated by cleaving plane from from its bulk crystalline form. The cleaving plane is described by the Miller Index notation (e.g. Si(111)). The cleavage plane (equivalent to termination plane) is a surface; all of its sites can be described by a unique 2D lattice. Therefore, interfacing film and substrate slabs geometrically implies the mapping of their respective 2D lattices. If the film and substrate lattices match, it is described as an epitaxial match, with a 2D superlattice that describes the interfaced lattice. This 2D superlattice contains a set of primative translation vectors (a,b) that describes both sides of the slab and their termination surface. Fig 1. below shows a schematic of how a new lattice is created at the interface of two slabs. Note: since it is a 2D representation, there is a 1D superlattice at the interface.
Figure 1. Lattice matching between Si(111) and AlO(101) faces. A cell made of 21 sapphire unit cells has almost exactly the same dimensions as a cell made of 40 silicon unit cells. This can be described by a new superlattice ('supervector') at the interface. Figure from Zur and McGill []
Finding the epitaxial lattice match between hetergenous interfaces implies finding a 2D superlattice that both sides must satisfy (or approximately satisfy). However, any interface can contain multiple sets of solutions to the primitive translation vectors (a,b) that still satisfies the 2D superlattice. As such, the goal is to look for the smallest possible values of the primative primitive translation vectors (a,b), also known as the reduced primitive translational vectors. The reduced primitive vectors has a unique solution for a,b, and α, unlike the general primitive translation vector set. Zurr and McGill proposed the following algorithm to find the reduced lattice set:
look for a being the shortest possible nonzero vector of the superlattice
look for being the shortest possible nonzero vector of the superlattice that is linearly independent of a
find angle α between vectors (a,b) that is non-obtuse.
The algorithm above is also shown in the flowchart in Fig 2. By leveraging computational resources and data from MP, it becomes possible to scan across all different cleavege planes for both the substrates and films to determine a set of reduced lattice planes, and therefore the epitaxial matches.
Fig 2. Flowchart of the unit cell reduction procedure. Figure from Zurr and McGill []
Most heterogenous interfaces will experience lattice mismatches. The following ratio describes the unit cell matching between the film and the substrate:
Where A1,A2corresponds to the unit cell areas of the original lattice of the film and substrate, and r1,r2 correspond to an integer value that satifies the unit cell areas being matched on the superlatice by the film and substrate. For lattice mismatches, we can set an upper limit for r1,r2by introducing Amax, where it must satify r1A1≈r2A2<Amax. And therefore:
The Suggest Substrates tool was first developed to study expitaxial polymorph stabilization through substrate selection [2]. This function is based upon the CoherentInterfaceBuilder function in pymatgen.
Author
Bryant Li
References
[1] A. Zur and T. C. McGill , "Lattice match: An application to heteroepitaxy", Journal of Applied Physics 55, 378-386 (1984) https://doi.org/10.1063/1.333084
[2] Hong Ding, Shyam S. Dwaraknath, Lauren Garten, Paul Ndione, David Ginley, and Kristin A. Persson ACS Applied Materials & Interfaces 2016 8 (20), 13086-13093 DOI: 10.1021/acsami.6b01630
r2r1=A1A2
r1<A1Amax,r2<A2Amax
2. Click on Apps
Step 2 screenshot
3. Click on Synthesis Explorer
Step 3 screenshot
4. Go to Search synthesis recipes
Step 4 screenshot
5. Type Target Formula or Keywords. We chose Li4Ti5O12.
Step 5 screenshot
6. Click on Search
Step 6 screenshot
7. There are 194 recipes for our search. We can narrow this down with Filters.
Step 7 screenshot
8. Go to Filters
Step 8 screenshot
9. Click on Materials and Keywords
Step 9 screenshot
10. We can add Precursor Material Formula and/or Keywords. We add TiO2 as a Precursor Material Formula.
Step 10 screenshot
11. Now there are 160 synthesis recipes. We can filter further with Synthesis Procedures.
Step 11 screenshot
12. Click on Synthesis Procedures
Step 12 screenshot
13. In Synthesis Procedures, we can choose various filters, such as Synthesis Type. Here, we chose Solid State Synthesis.
Step 13 screenshot
14. We can also change variables like Heating Temperature and Heating Time.
Step 14 screenshot
15. Applying these filters brings our search down to 107 synthesis recipes. We can look through these or continue to apply filters as desired.
How electronic band structures and density of states are calculated on the Materials Project (MP) website.
Calculation Details
A relaxed structure associated with the canonical data in the entries field of a material data entry is used to run both uniform and line-mode NSCF calculations with the same functional (and U if any). Currently, only GGA (PBE) and GGA+U DOS and band structures are available from the database.
We first run a static (SCF) calculation with a uniform (Monkhorst Pack or Γ-centered for hexagonal systems) k-point grid determined by the standard MPStaticSet input set in pymatgen. The charge density is extracted from this and used for the subsequent uniform and line-mode NSCF calculations. The parameters for both of these are determined by the MPNonSCFSet input set in pymatgen. For more details, see the band structure workflow in .
Line-mode Band Structure
The line-mode NSCF calculation is run with k-points chosen along high-symmetry lines within the Brillouin zone of the material. Currently, three conventions for choosing this k-path are used, and follow the methodologies by Curtarolo et al. , Hinuma et al. , and Munro et al. Code for generating the k-paths can be found within .
The Setyawan-Curtarolo band structure data is displayed on the website by default with full lines for spin-up and dashed lines for spin-down. For insulators, the band gap is computed according to the band structure. The nature of the gap (direct or undirect) as well as the k-points involved in the band gap transition are displayed. The VBM and CBMs are displayed for insulators as well by purple dots. Note that the website might not show all bands included in the calculation. These can be obtained by by downloaded the band structure data from the .
Density of States (DOS)
The DOS displayed on the website shows the total DOS, and elemental projections by default. However, total orbital and elemental orbital projections are also calculated and available from the . Please note that the DOS data and line-mode band structure may not completely agree on all derived properties such as the band-gap due to k-point grid differences. For instance, the uniform k-point grid used to calculate the DOS might not include some specific k-points along one of the high-symmetry lines, while the line-mode band structure will.
Material Band Gap
The band gap listed for a given material is chosen from one of its calculations. The current calculation hierarchy is as follows:
Density of States > Line-mode Band Structure > Static (SCF) > Optimization
Accuracy of Band Structures
Note: The term 'band gap' in this section generally refers to the fundamental gap, not the optical gap. The difference between these quantities is reported to be small in semiconductors but significant in insulators.
Figure 1: Experimental versus computed band gaps for 237 compounds in an internal test. The computed gaps are underestimated by an average factor of 1.6, and the residual error even after accounting for this shift is significant (MAE of 0.6 eV). We thank M. Chan for her assistance in compiling this data.
Density functional theory is formulated to calculate ground state properties. Although the band structure involves excitations of electrons to unoccupied states, the Kohn-Sham energies used in solving the DFT equations are often interpreted to correspond to electron energy levels in the solid.
The correspondence between the Kohm-Sham eigenvalues computed by DFT and true electron energies is theoretically valid only for the highest occupied electron state. The Kohn-Sham energy of this state matches the first ionization energy of the material, given an exact exchange-correlation functional. However, for other energies, there is no guarantee that Kohn-Sham eigenvalues will correspond to physical observables.
Despite the lack of a rigorous theoretical basis, the DFT band structure does provide useful information. In general, band dispersions predicted by DFT are reported to match experimental observations; one small test of band dispersion accuracy found that errors ranged from 0.1 to about 0.4 eV. However, predicted band gaps are usually severely underestimated. Therefore, a common way to interpret DFT band structures is to apply a 'scissor' operation whereby the conduction bands are shifted in energy by a constant amount so that the band gap matches known experimental observations.
Band gaps
In general, band gaps computed with common exchange-correlation functionals such as the LDA and GGA are severely underestimated. Typically the disagreement is reported to be around 50% in the literature. Some internal testing by the Materials Project supports these statements; typically, we find that band gaps are underestimated by about 40% (Figure 1). We additionally find that several known insulators are predicted to be metallic.
Origin of band gap error and improving accuracy
The errors in DFT band gaps obtained from calculations can be attributed to two sources: 1. Approximations employed to the exchange correlation functional 2. A derivative discontinuity term, originating from the true density functional being discontinuous with the total number of electrons in the system.
Of these contributions, (2) is generally regarded to be the larger and more important contribution to the error. It can be partly addressed by a variety of techniques such as the GW approximation but typically at high computational cost.
Strategies to improve band gap prediction at moderate to low computational cost now been developed by several groups, including Chan and Ceder (delta-sol), Heyd et al. (hybrid functionals) , and Setyawan et al. (empirical fits) . (These references also contain additional data regarding the accuracy of DFT band gaps.) The Materials Project may employ such methods in the future in order to more quantitatively predict band gaps. For the moment, computed band gaps should be interpreted with caution.
Citation
To cite the calculation methodology, please reference the following works:
A. Jain, G. Hautier, C. Moore, S.P. Ong, C.C. Fischer, T. Mueller, K.A. Persson, G. Ceder., A High-Throughput Infrastructure for Density Functional Theory Calculations, Computational Materials Science, vol. 50, 2011, pp. 2295-2310.
Authors
Anubhav Jain
Shyue Ping Ong
Geoffroy Hautier
References
[1]: W. Setyawan, S. Curtarolo, High-throughput electronic band structure calculations: Challenges and tools, Computational Materials Science 2010, 49, 299-312.
[2]: Y. Hinuma, P. Giovanni, Y. Kumagai, F. Oba, I. Tanaka, Band structure diagram paths based on crystallography Computational Materials Science 2017, 128, 140-184.
[3]: J.M. Munro, K. Latimer, M.K. Horton, S. Dwaraknath, K.A. Persson, An improved symmetry-based approach to reciprocal space path selection in band structure calculations, npj Computarional Materials 2020, 6, 112.
[4]: E.N. Brothers, A.F. Izmaylov, J.O. Normand, V. Barone, G.E. Scuseria, Accurate solid-state band gaps via screened hybrid electronic structure calculations., The Journal of Chemical Physics. 129 (2008)
[5]: R. Godby, M. Schluter, L.J. Sham, Self-energy operators and exchange-correlation potentials in semiconductors, Physical Review B. 37 (1988).
[6]: M. Chan, G. Ceder, Efficient Band Gap Predictions for Solids, Physical Review Letters 19 (2010)
[7]: J. Heyd, J.E. Peralta, G.E. Scuseria, R.L. Martin, Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional, Journal of Chemical Physics 123 (2005)
[8]: W. Setyawan, R.M. Gaume, S. Lam, R. Feigelson, S. Curtarolo, High-throughput combinatorial database of electronic band structures for inorganic scintillator materials., ACS Combinatorial Science. (2011).
Piezoelectric Constants
How piezoelectric constants are calculated for the Materials Project (MP) website.
Introduction
Piezoelectricity is a reversible physical process that occurs in some materials whereby an electric moment is generated upon the application of a stress. This is often referred to as the direct piezoelectric effect. Conversely, the indirect piezoelectric effect refers to the case when a strain is generated in a material upon the application of an electric field. The mathematical description of piezoelectricity relates the strain (or stress) to the electric field via a third order tensor. This tensor describes the response of any piezoelectric bulk material, when subjected to an electric field or a mechanical load.
The piezoelectric constants from the Materials Project (MP) are calculated from first principles Density Functional Perturbation Theory (DFPT) [1] and are approximated as the superimposed effect of an electronic and ionic contribution. From the full piezoelectric tensor, several properties are derived such as the maximum longitudinal piezoelectric modulus and the corresponding crystallographic direction. Just as with the elastic constants, multiple consistency checks are performed on all the calculated piezoelectric data to ensure its reliability and accuracy.
Figure 1: longitudinal piezoelectric modulus-surface for a cubic compound, showing the maximum response in the <111> family of directions.
Formalism
In this work, we calculate the piezoelectric stress coefficients, from DFPT, with units of . These can be defined in terms of thermodynamic derivatives as shown below .
where , , , and represent the electric displacement field, the electric field, the strain tensor, the stress tensor and the temperature, respectively.
The above relations can be written in Voigt-notation as shown below.
We note that the most commonly used piezoelectric constants appearing in the (experimental) literature are the piezoelectric strain constants, usually denoted by . These can be readily related to the constants if the elastic compliances (at constant electric field and temperature) of the materials are known: . In particular, the piezoelectric strain constants can be expressed thermodynamically as shown below
It is well-known that the piezoelectric behavior can only occur in crystals that lack inversion symmetry. This is the direct consequence of the symmetry properties of the piezoelectric tensor, which is of order 3. Another fundamental requirement for piezoelectric behavior is that the material has a band gap. Combined, these criteria severely limit the amount of compounds in nature that have the potential to exhibit piezoelectric behavior.
For the Materials Project in particular, potential piezoelectric materials in the database are identified by i) allowing only structures with space groups 1, 3-9, 16-46, 75-82, 89-122, 143-146, 149-161, 168-174, 177-190, 195-199, 207-220 (since these space groups lack inversion symmetry), and in addition ii) the calculated DFT bandgap of the material > 0.1 eV. Compounds in the Materials Project database that satisfy these criteria are selected for a full-DFT calculation of the piezoelectric tensor and derived properties (see below).
Derived piezoelectric properties
For elastic properties, which are based on a tensor of order 4, isotropic Voigt and Reuss averages can be derived on the bulk and shear moduli. For piezoelectric properties, this isotropic averaging-approach does not quite work due to the requirement that inversion symmetry cannot occur in piezoelectric materials. On MP, in addition to the piezoelectric tensor in Voigt-notation, we report the maximum longitudinal piezoelectric modulus of the compound and the corresponding crystallographic direction in which this occurs. One can think of these quantities as the piezoelectric counterpart of the well-known Young's modulus and the stiffest elastic direction in the context of elasticity-theory. Fig. 1 shows an example of how the longitudinal piezoelectric modulus can be represented in 3D. This is for the case of a cubic material. As can be seen clearly, the maximum modulus occurs in the <111> family of crystallographic directions. By symmetry, this is always the case for cubic piezoelectric materials. Fig. 2 shows a more complicated longitudinal piezoelectric modulus-surface for an orthorhombic compound. In that case, the relative magnitudes of the tensor components dictate in which crystallographic direction, the maximum response occurs. Finally, note that for some compounds, a piezoelectric response is only induced by shear deformation rather than tensile or compressive deformation. For these cases, the response cannot be depicted such as in Figs. 1 and 2. The representations such as in Figs. 1 and 2 and created using the open-source MTEX package .
Figure 2: longitudinal piezoelectric modulus-surface for an orthorhombic compound.
DFT parameters
The first-principles results presented in this work are performed using the projector augmented wave (PAW) method as implemented in the Vienna Ab Initio Simulation Package (VASP). In all calculations, we employ the Perdew, Becke and Ernzerhof (PBE) Generalized Gradient Approximation (GGA) for the exchange-correlation functional. A cut-off for the plane waves of 1000 eV is used and a uniform k-point density of approximately 2,000 per reciprocal atom (pra) is employed, which means that the number of atoms per cell multiplied by the number of k-points equals approximately 2,000. For the compounds that contain magnetic elements, a ferromagnetic state is initialized in the calculation. Similarly to our previous work, we expect to correctly converge to ferromagnetic and non-magnetic states in this way, but not to anti-ferromagnetic states. Due to the presence of strongly correlated electrons in some of the oxides, the GGA+U method is employed, with U representing the Hubbard-parameter. The values of U are chosen consistent with those employed in MP.
Figure 3: A graphical representation of the piezoelectric dataset, currently containing over 900 materials. A series of concentric circles indicate constant values of the maximum longitudinal piezoelectric modulus, . The compounds are broken up according to the crystal system and the different point group symmetry-classes considered in this work. See the paper for details.
Crystal symmetry
The crystal symmetry and in particular the point group dictates the symmetry of the piezoelectric tensor, relates components of the tensor to each other and imposes that certain components equal zero. All piezoelectric tensors in the Materials Project have been symmetrized for consistency with the underlying point group of the compound. Figure 4 gives an overview of the symmetrized piezoelectric tensors in MP, broken up by the different piezoelectric point groups. Also, typical surface representations are shown. The point group that only yields piezoelectric behavior upon the application of shear is not included in the representation in Fig. 4.
Figure 4: Piezoelectric tensors and symmetry classes considered in this work. Typical representations of the longitudinal piezoelectric modulus in 3D are also shown for each crystal point group. Note that depending on the components of the piezoelectric tensor, the surface representation can differ from those shown here. See the paper for details.
Citation
To cite the piezoelectric properties within the Materials Project, please reference the following work:
de Jong, Maarten and Chen, Wei and Geerlings, Henry and Asta, Mark and Persson, Kristin Aslaug. A database to enable discovery and design of piezoelectric materials,
The paper presents the results of our piezoelectric constant-calculations for the first batch of 941 compounds. Our DFT-parameters, the workflow and comparison to experiments are described in detail. Also, the filters in the workflow used for detecting anomalies in the calculations are described in the paper.
Authors
Maarten de Jong
References
[1]: Baroni, Giannozzi S. P. and Testa, A. Phys. Rev. Lett. 58, 1861 (1987)
[2]: Nye, J. F. Physical properties of crystals (Clarendon press, 1985).
[3]: Bachmann, F., Hielscher, R. & Schaeben, H. Texture analysis with MTEX-free and open source software toolbox. Solid State Phenomena 160, 63–68 (2010).
[4]: Hielscher, R. & Schaeben, H. A novel pole figure inversion method: specification of the MTEX algorithm. Journal of Applied Crystallography 41, 1024–1037 (2008).
[5]: Mainprice, D., Hielscher, R. & Schaeben, H. Calculating anisotropic physical properties from texture data using the MTEX open-source package. Geological Society, London, Special Publications 360, 175–192 (2011).
Structure Sources
Where did each of the initial structures come from?
Each material in the QMOF Database, and thereby the MOF Explorer, was taken from an existing dataset of MOF structures. Some of these datasets are dedicated to experimentally synthesized MOF structures, whereas others are hypothetical MOF structures (i.e. computationally constructed). Below, we outline the various datasets of MOF structures used in constructing the QMOF Database.
Cambridge Structural Database – MOF Subset
The Cambridge Structural Database (CSD) contains experimentally derived crystal structures for over a million materials. Of the crystal structures published on the CSD, approximately 100,000 are included in what is referred to as the CSD MOF Subset. It should be noted that the definition of a MOF in the CSD MOF Subset is more inclusive than many other databases and includes non-porous materials that are arguably best described as coordination polymers, in addition to more conventional MOF structures.
In the QMOF Database, structures were taken directly from the CSD MOF Subset with free (i.e. unbound) solvent removed from the pores. was used to download the structures, and we excluded materials that were flagged as having charge-balancing ions, any errors in the crystal structure, or disorder in the framework. Additionally, we excluded any structures that lacked carbon or hydrogen atoms, had atoms with close interatomic distances, had lone (i.e. unbonded) atoms, or had terminal oxo ligands on metals where such ligands are typically OH groups or water. Several scripts to carry out these fidelity checks can be found .
CoRE MOF Database
The contains experimentally derived crystal structures for ~14,000 porous, three-dimensional MOFs. The materials in the CoRE MOF Database were derived from the CSD but are not directly associated with the CSD MOF Subset, although many of the CoRE MOFs can be found in the CSD MOF Subset as well. Unlike the CSD MOF Subset, which provides as-reported crystal structures, a suite of automated and manual structural corrections were carried out during the construction of the CoRE MOF Database. As with any automated approach, not all of these structural corrections are perfect in their execution and can result in materials with misplaced atoms, under- and over-bonded atoms, charge imbalances, and similar structural fidelity issues that can be determinetal for DFT.
In the QMOF Database, we considered CoRE MOFs that were included in curated lists provided by and to increase the likelihood of having high-fidelity CoRE MOF structures. For consistency, the free solvent-removed (FSR) subset of the CoRE MOF Database was conisdered. We emphasize that there are many MOFs present in the CoRE MOF Database that we instead adopted from the CSD MOF Subset directly. As such, if a user is specifically interested in which MOFs in the QMOF Database are also present in the CoRE MOF Database, one should compare the CSD reference codes and/or MOFids for the materials in these two datasets.
Pyrene MOFs
Several experimentally characterized, pyrene-containing MOFs were taken from the work of using the structures that were uploaded to the . No further modifications were made to these structures.
ToBaCCo
The can generate hypothetical MOFs from known inorganic and organic building blocks (and topologies). Here, the "ToBaCCo" dataset of MOFs specifically refers to those found in the by Colón, Gómez-Gualdrón, and Snurr. In the QMOF Database, MOFs with triangular Cu-containing nodes were selected from the ToBaCCo dataset, as found .
Anderson and Gómez-Gualdrón
The contains hypothetical MOFs constructed using ToBaCCo. In the QMOF Database, we selected Zr-containing MOFs from this dataset. We also expanded the dataset to include hypothetical Hf-containing MOfs by exchanging the Zr species for Hf.
Boyd & Woo
Hypothetical MOFs in the QMOF Database were also adopted from the work of using the dataset of structures uploaded to the Materials Cloud . These MOFs were construced using the , as described in prior work by . As a result, we refer to these hypothetical MOFs as coming from the Boyd & Woo dataset.
In the QMOF Database, we adopted MOFs from select families in the Boyd & Woo daaset and occasionally made modifications to several of these MOFs to diversify our collection. For instance, we occasionally exchanged the metals in the inorganic node, and we constructed Al rod MOFs by exchanging the metals in the pre-existing V rod MOFs and protonating the bridging oxo ligands. We still refer to these structures as being derived from the Boyd & Woo dataset even though custom modifications have been made.
Genomic MOF Database
Hypothetical MOFs from the made available were included in the QMOF Database. These structures were adopted as-is without further modification.
Mail-Order MOF-5s
Hypothetical MOF-5 analogues were obtained from by Haranczyk and colleagues. See for the dataset.
Hypothetical MOF-74s
Hypothetical Mg-MOF-74 analogues were obtained from by Haranczyk and colleagues.
How dielectric constants are calculated on the Materials Project (MP) website.
Introduction
A dielectric is a material that can be polarized by an applied electric field. This limits the dielectric effect to materials with a non-zero band gap. The mathematical description of the dielectric effect is a tensor constant of proportionality that relates an externally applied electric field to the field within the material. Along with the elastic and piezoelectric tensors, the dielectric tensor provides all the information necessary for the solution of the constitutive equations in applications where electric and mechanical stresses are coupled.
The dielectric tensors from the Materials Project (MP) are calculated from first principles Density Functional Perturbation Theory (DFPT) and are approximated as the superimposed effect of an electronic and ionic contribution. From the full piezoelectric tensor, several properties are derived such as the refractive index and potential for ferroelectricity. Just as with the piezoelectric and elastic constants, multiple consistency checks are performed on all the calculated dielectric data to ensure its reliability and accuracy.
Formally, the dielectric tensor ε relates the externally applied electric field to the field within the material and can be defined as:
where E is the electric field inside the material and E0is the externally applied electric field. the indices i,j refer to the direction in space and take the values: 1,2,3. The dielectric tensor can be split in the ionic (ϵ0) and electronic (ϵ∞) contributions:
Here, we consider only the response of non-zero band gap materials to time-invariant fields. In the hypothetical case that a material does not respond at all to the external field, ϵij∞ would be equal to the identity tensor and ϵij0 would be zero. In fact, materials with zero ionic contribution do exist. In general, for ϵij0 to be non-zero, compounds need to have at least 2 atoms per primitive cell, each having a different atomic charge. The dielectric tensor is symmetric and respects all the symmetry operations of the corresponding point group. This limits the number of independent elements in the tensor to a minimum of 1 and a maximum of 6 depending on the crystal symmetry.
The dielectric response calculated herein corresponds to that of a single crystal. In polycrystalline samples, grains are oriented randomly and hence, the actual response will be different. Generally, the dielectric response varies with the frequency of the applied external field however here, we consider the static response (i.e., the response at constant electric fields or the long wavelength limit). Since the ionic contribution vanishes at high frequencies, our results can be used to obtain an estimate of the refractive index, n, at optical frequencies and far from resonance effects using the well known formula: [2]
where ϵpoly∞ is the average of the eigenvalues of the electronic contribution to the dielectric tensor. It should be noted this equation for the refractive index assumes the material is non-magnetic.
DFT Parameters
The initial set of 1,056 dielectric tensors were calculated using the Vienna Ab-Initio Simulation Package [3-6] (VASP version 5.3.4) combined with the Generalized Gradient Approximation GGA/PBE[7,8]+U[9,10] exchange-correlation functional and Projector Augmented Wave pseudopotentials [11,12]. The U values are energy corrections that address the spurious self-interaction energy introduced by GGA. Here, we used U values for d orbitals only that were fitted to experimental binary formation enthalpies using Wang et al. [13] method. The full list of U values used, can be found in ref.[10]. The k-point density was set at 3,000 per reciprocal atom and the plane wave energy cut-off at 600 eV (ref. 4). For detailed information on the calculation of the dielectric tensor within the DFPT framework we refer to Baroni et al. [14,15] and Gonze & Lee [16].
Piezoelectricity calculations use the same DFPT methodology with a tighter parameter set to achieve convergence. As a result, the dielectric tensor is already converged in these calculations and is reported for any non-centrosymmetric material, not in the initial dataset of dielectrics.
Benchmarking
We see that in most cases, it is possible to predict the dielectric constant of materials with a relative deviation of less than +/−25% from experimental values at room temperature. Including local field effects gives the smallest mean absolute relative deviation ( MARD= 16.2 % for GGA). Furthermore, we note a tendency to overestimate rather than underestimate the dielectric constant relative to experiments, which is a well-known effect of DFPT [17,18,19] for the electronic contribution. Although it has often been related to the band gap underestimation problem of DFT, DFPT is a ground state theory and hence, the dielectric constant should, in principle, be described exactly [20]. In fact, as described by various authors, the problem is likely linked to the exchange-correlation functional [21-26]. Specifically, the exchange correlation functional has been found to depend on polarization but the actual dependence formula is, unfortunately, not known [27,28]. Additionally, the validity of GGA depends on the charge density varying slowly—an assumption that may be broken when an external electric field is applied [30].
Citation
To cite the dielectric properties within the Materials Project, please reference the following work:
Benchmarking density functional perturbation theory to enable high-throughput screening of materials for dielectric constant and refractive index. Ioannis Petousis, Wei Chen, Geoffroy Hautier, Tanja Graf, Thomas D. Schladt, Kristin A. Persson, and Fritz B. Prinz. Phys. Rev. B 93(11). DOI:10.1103/PhysRevB.93.115151
High-throughput screening of inorganic compounds for the discovery of novel dielectric and optical materials. Ioannis Petousis, David Mrdjenovich, Eric Ballouz, Miao Liu, Donald Winston, Wei Chen, Tanja Graf, Thomas D. Schladt, Kristin A. Persson, and Fritz B. Prinz. Scientific Data 4. DOI:10.1038/sdata.2016.134
These papers present the results of our dielectric constant-calculations for the first batch of 1,056 compounds. Our DFT-parameters, the workflow, the workflow filters used for detecting anomalies in the calculations and comparison to experiments are described in detail.
Authors
Shyam Dwaraknath
Ioannis Petousis
References
[1]: Baroni, Giannozzi S. P. and Testa, A. Phys. Rev. Lett. 58, 1861 (1987)
[2]: Petousis I. et al. Benchmarking of the density functional perturbation theory to enable the high-throughput screening of materials for the dielectric constant and refractive index. Phys. Rev. B 93, 115151 (2016).
[3]: Kresse G. & Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
[4]: Kresse G. & Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251 (1994).
[5]: Kresse G. & Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
[6]: Kresse G. & Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
[7]: Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
[8]: Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396 (1997).
[9]: Dudarev S. L., Botton G. A., Savrasov S. Y., Humphreys C. J. & Sutton A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505 (1998).
[10]: Jain A. et al. A high-throughput infrastructure for density functional theory calculations. Comp. Mater. Sci. 50, 8 2295 (2011).
[11]: Blöchl P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
[12]: Kresse G. & Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
[13]: Wang L., Maxisch T. & Ceder G. Oxidation energies of transition metal oxides within the GGA+ U framework. Phys. Rev. B 73, 195107 (2006).
[14]: Baroni S., Giannozzi P. & Testa A. Elastic constants of crystals from linear-response theory. Phys. Rev. Lett. 59, 2662 (1987).
[15]: Baroni S., de Gironcoli S., Dal Corso A. & Giannozzi P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
[16]: Gonze X. & Lee C. Dynamical matrices, born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 55, 10355 (1997).
[17]: N. Marzari and D. J. Singh, Phys. Rev. B 62, 12724 (2000). [^18]: A. Dal Corso, S. Baroni, and R. Resta, Phys. Rev. B 49, 5323 (1994).
[19]: F. Kootstra, P. L. de Boeij, and J. G. Snijders, Phys. Rev. B 62, 7071 (2000).
[20]: A. Dal Corso, S. Baroni, and R. Resta, Phys. Rev. B 49, 5323 (1994).
[21]: A. Dal Corso, S. Baroni, and R. Resta, Phys. Rev. B 49, 5323 (1994).
[22]: V. Olevano, M. Palummo, G. Onida, and R. Del Sole, Phys. Rev. B 60, 14224 (1999).
[23]: W. G. Aulbur, L. Jönsson, and J. W. Wilkins, Phys. Rev. B 54, 8540 (1996).
[24]: Ph. Ghosez, X. Gonze, and R. W. Godby, Phys. Rev. B 56, 12811 (1997).
[25]: R. Resta, Phys. Rev. Lett. 77, 2265 (1996). [^26]: R. Resta, Phys. Rev. Lett. 78, 2030 (1997). [27]: A. Dal Corso, S. Baroni, and R. Resta, Phys. Rev. B 49, 5323 (1994).
[28]: W. G. Aulbur, L. Jönsson, and J. W. Wilkins, Phys. Rev. B 54, 8540 (1996).
[29]: Ph. Ghosez, X. Gonze, and R. W. Godby, Phys. Rev. B 56, 12811 (1997).
[30]: V. Olevano, M. Palummo, G. Onida, and R. Del Sole, Phys. Rev. B 60, 14224 (1999).
Recent list of Materials Project documentation contributors.
Database Versions
A changelog of Materials Project (MP) database releases.
This page contains a summary of major changes for each version of the Materials Project database.
We are aware of a community need for more detailed change logs, and hope to improve our reporting for future database versions.
Database versions are labelled via the date they become generally available to the public. For advanced users, the public database label is mapped to an internal pull request number, and this is accessible in the API via the builder_meta key.
Anion and GGA/GGA+U Mixing
Details on anion and GGA/GGA+U mixing scheme corrections
This correction scheme assumes independent, linear corrections associated with each corrected element. For example, would receive both a '' and an 'oxide' correction (as explained below), while elemental would receive no corrections. For complete details of our correction scheme, refer to Wang et al.
Methodology
Equations of State (EOS)
How equations of state (EOS) are calculated on the Materials Project (MP) website.
Introduction
Thermodynamic equations of state (EOS) for crystalline solids describe material behaviors under changes in pressure, volume, entropy and temperature. Despite over a century of theoretical development and experimental testing of energy-volume (E-V) EOS for solids, there is still a lack of consensus with regard to which equation is optimal, as well as to what metrics are most appropriate for making this judgment.
Calculation of EOS is automated using self-documenting workflows compiled in the atomate code base. Atomate couples pymatgen for materials analysis, custodian for just-in-time debugging of DFT codes, and Fireworks for workflow management. The EOS workflow begins with a structure optimization and subsequently calculates the energy of isotropic deformations including ionic relaxation with volumetric strain ranging from -15.7% to 15.7% (-5% to 5% linear strain) of the optimized structure. Density-functional-theory (DFT) calculations were performed as necessary using the projector augmented wave (PAW) method as implemented in the Vienna Ab Initio Simulation Package (VASP) within the Perdew-Burke-Enzerhof (PBE) Generalized Gradient Approximation (GGA) formulation of the exchange-correlation functional. A cut-off for the plane waves of 520 eV is used and a uniform k-point density of approximately 1,000 per reciprocal atom is employed. In addition, standard Materials Project Hubbard U corrections are used for a number of transition metal oxides, as documented and implemented in the pymatgen VASP input sets. We note that the computational and convergence parameters were chosen consistently with the settings used in the Materials Project to enable direct comparisons with the large set of available MP data.
Parameters and Convergence
Parameter and convergence details for r2SCAN calculations run by the Materials Project
Calculation Parameters
We use the projector-augmented wave (PAW) or modeling core electrons with an energy cutoff of 680 eV. K-point grids were generated automatically by VASP using KSPACING values ranging from 0.22/Å to 0.44/Å. Specifically, the Monkhorst-Pack method is used for grid generation (with -centered for hexagonal cells), and the tetrahedron method is used to perform the k-point integrations. These were determined from the GGA-estimated bandgap of each material based on the work by Wisesa et al. . More details regarding the calculation method can be found in ref ; however, the Materials Project has updated many parameters as documented throughout the Methodology sections. The most up-to-date input sets can be .
You can verify the current database version powering the website on the footer of every page. If you are using the API, there is a get_database_version method available.
v2022.10.28
This database build incorporates Materials Project’s (R2)SCAN calculations as pre-release data. The default fields returned by the website and API will remain unchanged from the previous release at the GGA(+U) level of theory, but the (R2)SCAN data is now available for advanced users. Either see the “Pre-release Data” section of a relevant material details page, generate an R2(SCAN) phase diagram with the Phase Diagram app, or access the data via the thermo API endpoint. This database release also incorporates several new perovskite materials from a collaboration with Zachary Bare, University of Colorado.
v2021.11.10
This will be the first release with our new website and API. It does not contain any new data but is built using our new database building methods and is largely consistent with the previous database release. Some changes exist to the previous release due to improvements to detection of multi-anion systems leading to changes in the applied formation energy corrections.
Be aware, database version v2021.11.10 onwards is only available on the new Materials Project website and API. The legacy website and legacy API are frozen to the v2021.05.13 database release.
v2021.05.13
This release updates the energy correction scheme we use to generate phase diagrams and compute formation energies. As with any new database release, formation energies for many compounds have changed; however in this case the change is due only to our new energy correction scheme and not to any new data. We are proud to report that the new correction scheme has reduced the overall error in formation energy in our database by 7% compared to experiment.
You can see details of each correction that has been applied by inspecting the energy_adjustments attribute of a ComputedEntry retrieved via the API. In addition, the new correction scheme is available for manual use via the MaterialsProject2020Compatibility class in pymatgen.
We realize that this change may be disruptive to ongoing work, and want to assure you that the historical corrections are still available in pymatgen if needed. They may be recovered by manually reprocessing ComputedEntry using the legacy MaterialsProjectCompatibility class. An example notebook demonstrating how to do this available on matgenb 25.
Below we summarize the most significant changes associated with the new MaterialsProject2020Compatibility correction scheme. For complete details and documentation, please refer to this manuscript 32.
1. Refitted corrections for legacy species
Corrections applied to oxygen compounds, diatomic gases, and transition metal oxides and fluorides have been refit using more up to date DFT calculations and a larger compilation of computed and experimental formation enthalpy data.
2. Corrections for additional species
We have added corrections for Br, I, Se, Si, Sb, and Te, which did not previously have energy corrections. As a result, formation energies for materials containing these species will generally be lower than they were previously.
3. Diatomic gas corrections moved to compounds
Previously, corrections for H, F, Cl, and N were applied to the elements. One consequence of this was that polymorphs of H2, N2, Cl2 and F2 were alwaysassigned a zero energy above hull, even if some polymorphs were higher in energy. This made interpretation of these values confusing. With this release, energy corrections are applied to the material (e.g., LiH) and not the element. This also means that unstable polymorphs of diatomic gases will now have non-zero e_above_hull
4. Oxidation state based corrections
Our build process now estimates the likely oxidation states of each species in a material, and uses this information to intelligently apply corrections to anionic species only when their estimated oxidation state is negative. For example, in the compound MoCl3O, estimated oxidation states for both Cl and O are negative, so both anions receive corrections.
Our algorithms are not always successful in predicting the oxidation state. When this occurs, we apply anion corrections to only the most electronegative element in the material. As a result, some ternary or higher compounds in the database may be destabilized in this release because their oxidation states could not be determined. This is the case for MoCl5O (mp-1196724) for example, which does not receive a Cl correction because O is more electronegative.
If this affects your work, you can manually assign oxidation states by populating the oxidation_states key of the .data attribute of any ComputedEntry and then reprocessing the data using MaterialsProject2020Compatibility.
5. Uncertainty Quantification
We now compute the estimated uncertainty associated with the energy corrections on a material. Uncertainties reflect the measured uncertainty in the underlying experimental data that we use to determine the corrections, as well as uncertainty associated with the fitting procedure itself. This information enables new methods of assessing phase stability, as described in this manuscript 32
A Note for API and MPRester Users
For API users, if you are retrieving formation energies directly via the API, you will get the correct, latest formation energies from the current database release. However, if you are using get_entries or get_pourbaix_entries which apply the correction scheme on-the-fly, make sure to update to the latest version of pymatgen (v2022.0.8 or later) to get the correct values. If you are using pymatgen v2021 or earlier, this will use the old correction scheme by default when using get_entries and get_pourbaix_entries.
v2021.03.22
This release updates some older materials with new calculations, and adjusts our rules for deprecating older calculations. It does not contain any new materials. Thanks to the new calculations many materials that were previously deprecated are now accessible again. This release is in preparation for a switch to our new compatibility scheme which will improve our predictions of formation energy.
v2021.02.08
We had a small new database release today, this introduces new higher-quality calculations for around 30,000 materials. It also deprecates 78 materials since we currently do not have calculations for these materials that match our current quality standards; we hope to restore these 78 materials in a subsequent release. For an exact list, please see the attached file.
As a reminder, all historical calculation tasks remain available via our API and the task detail pages, and information on deprecated materials also remain available via the API. More information on our deprecation policy is in our documentation. We continue our work on better ways communicate database diffs and to more easily provide access to historical information, so stay tuned for future announcements here.
This releases addresses issues noticed in the previous release with formation energies and updates the energies of approximately 6k materials where this error was greatest. We are planning a further supplemental release.
We’re also looking at ways to put in place a process to be more transparent with database changes and updates to share more specifically what has changed, as well as providing means to access historical versions of the database, since we know this is a common requirement.
Note that, wherever possible, we continue to keep individual historical calculation data available via its task_id even in cases where the aggregated information (such as that presented on the materials detail page) might change.
V2020.08.20 Released
In this release we have added thousands of new band structure and density of states calculations, improving our overall material coverage and data quality. Additionally, we have overhauled the plotting for these quantities on the material details page. This is a first step in improving the electronic structure data within the Materials Project as part of our new tool set 46 for band structure calculations.
We are also working through an on-going issue affecting the energies of a small number of materials. In the previous release, we added a large batch of higher-quality calculations for our energetics as well as fixing numerous bugs. However, we discovered an error in our calculation parameters leading to larger energies than expected for a minority of materials and issues such as those discussed here 27. We are currently re-running these calculations and will be fixing this data in a supplemental update in the next few weeks. We advise anybody performing large screening studies to do so with caution or wait until this supplemental update has been released.
v2020.06
In this database release, we have added several thousand materials and many magnetic ground states, improved the quality of our energetics, and fixed many bugs. This database release is part of on-going efforts in 2020 to improve database reliability and quality, following the introduction of our deprecation process last year. There are still known issues with this release which we are working to address, please let us know if you encounter any in our forum.
v2019.12.05
The issue mentioned in 2019-12-04 has now been addressed, however approximately 7% of materials saw errors in their reported energies above hull of greater than 0.05 eV/atom. Values calculated via pymatgen or via the phase diagram app on the website during this time were correct, while values reported on the materials details page and via the e_above_hull API key were incorrect.
We encourage users who accessed convex hulls from the website between the latest database release and 2019-12-05 to re-check any values obtained from the website.
We apologize for the error, and will be incorporating additional checks into our automated testing to prevent similar errors in the future.
v2019.12.04
We are aware of an on-going issue with the reported energies above hull on the materials detail pages. We will update this thread when a fix has been fully implemented and with further details.
Until this issue is fully resolved, correct energies above hull can be retrieved using pymatgen as follows:
If you have not previously used pymatgen, it is a Python code and can be installed using pip install pymatgen or conda install --channel conda-forge pymatgen.
Note: the above information for v2019-12-04 is now out of date.
v2019.11.21
During deployment of the new v2019.11 database, there was temporary issue with generating interactive phase diagrams leading to incorrect formation enthalpies for a small number of chemical systems. This has now been fixed. Data presented on the materials detail pages was unaffected by this issue.
v2019.11
Introduced 3,971 new materials
Amorphous materials added with amorphous tag
Added theoretical which is True when the material matches no known experimental structure from ICSD
Fixed several inconsistency bugs for band_gap, piezo tensors, elastic warnings, and total magnetic moment.
v2019.05
Introduced a new deprecated field to materials. By default the website and API only search for materials that are not deprecated: {“deprecated”: false}.
Deprecated 15,000 and added 3,600 new materials. We will be recomputing the deprecated materials to fill these spaces back up. Some of these new relaxations may end up matching current materials, so the total number of materials is not guaranteed to be the same as in V2019.02.
Fixed an issue with sandboxes not properly building the whole hull. Previously, only the sandboxed chemical systems were being recalculated for energy_above_hull searches
v2019.02
Added over 47,000 new materials from orderings of disordered ICSD as well as compounds from the Pauling File
Finalized enforcing symmetry on piezo tensors
Moved third order elastic data to elasticity_third_order so that people are not swamped by the mountain of information associated with it.
v2018.12
Adjusted the mp-id naming scheme to fix “mvc” ids taking over old mp-ids.
Fixed piezoeletric max_direction to be a miller index rather than a unit vector.
v2018.11
Changed the grouping of magnetic materials to aggregate all magnetic orderings of a given material into a single material-id, and report the lowest energy ordering
Fixed incorrect calculation and display of polycrystalline dielectric constants
Fixed labeling of all materials as high-pressure. Note we’re parsing ICSD tags for this labeling so while some materials may not conventionally be considered high-pressure, a single matching ICSD entry can tag a material as such. We would love to hear comments on how we could better tag high-pressure materials
Begun enforcing the symmetry of the structure on piezo tensors. In general, this reduces the expected piezo value.
from pymatgen import MPRester
from pymatgen.analysis.phase_diagram import PhaseDiagram
with MPRester(YOUR_API_KEY_HERE) as mpr:
# replace with your elements and mp-id of interest
entries = mpr.get_entries_in_chemsys(['Li','Co', 'O'])
entry_of_interest = mpr.get_entry_by_material_id('mp-19128')
phase_diagram = PhaseDiagram(entries)
e_above_hull = phase_diagram.get_e_above_hull(entry_of_interest)
print("e_above_hull", e_above_hull)
1) Anion corrections
For many elements that take on negative oxidation states in solids, differences in electron localization between the elements and the solid can result in substantial errors in formation energies computed from DFT calculations. This is especially true for elements that are gaseous in their standard state - O2, N2, Cl2, F2, and H2.
To address this, we adjust the energies of materials containing certain elements by applying a correction to anionic species, as explained in ref [1]. Specifically, we apply energy corrections to 14 anion species -- 'oxide', 'peroxide', 'superoxide', S, F, Cl, Br, I, N, H, Se, Si, Sb, and Te. In the case of oxygen-containing compounds, separate corrections are applied to oxides, superoxides, and peroxides based on the specific bonding environment of oxygen in the material, as determined from nearest-neighbor bond lengths (e.g., <1.35 Å for superoxide, <1.49 Å for 'peroxide', and 'oxide' otherwise). Thus, Na2O receives an 'oxide' correction while NaO2 receives a `superoxide' correction.
Anion corrections are applied to a material only when it contains a corrected element as an anion. For example, the 'H' correction is applied to LiH but not to H2O. A specie is classified as an anion if its estimated oxidation state (when available) is negative, or if it is the most electronegative element in the formula.
2) GGA / GGA+U Mixing Corrections
Some compounds are better modeled with a U correction term to the density functional theory Hamiltonian while others are better modeled without (i.e., regular GGA). Energies from calculations with the +U correction are not directly comparable to those without. To obtain better accuracy across chemical systems, we use GGA+U when appropriate, GGA otherwise, and mix energies from the two calculation methodologies by adding an energy correction term to the GGA+U calculations to make them comparable to the GGA calculations.
Specifically, we use GGA+U for oxide and fluoride compounds containing any of the transition metals V, Cr, Mn, Fe, Co, Ni, W, and Mo, and GGA for everything else. More details on this method can be found in refs. [1,2]
Accuracy of Total Energies
To estimate the accuracy of our total energy calculations, we compute reaction data and compare against experimental data. Note that this data set was compiled using a lower k-point mesh and pseudopotentials with fewer electrons than the current Materials Project parameter set.
Estimating errors in calculated reaction energies
The accuracy of calculated reaction energies depends on the chemical system investigated. In general, GGA calculations have similar errors among chemically similar systems. Hence, reaction energies between chemically similar systems (e.g., a reaction where the reactants and products are all oxides, such as MgO + Al2O3→MgAl2O4 tend to have smaller errors than reactions between chemically dissimilar systems (e.g., between metals and insulators).
Figure 1: Errors in Calculated Formation Energies for 413 binaries in the Kubaschewski Tables. Energies are normalized to per mol atom.
To provide a quantitative indicator of the error we may expect from the reaction calculator, we have computed the reaction energies of 413 binaries in the Kubaschewski Tables formed with Group V, VI and VII anions. Figure 1 shows the errors in the calculated formation energies (compared to the experimental values) for these compounds. The mean absolute error (MAE) is around 14 kJ mol−1. 75% of the calculated formation energies are within 20 kJ mol−1. We also found that compounds of certain elements tend to have larger errors. For example, Bi, Co, Pb, Eu, U, Tl and W compounds often have errors larger than 20 kJ mol−1.
It should be noted that while an MAE of 14 kJ mol−1 is significantly higher than the desired chemical accuracy of 4 kJ mol−1, it compares fairly well with the performance of most quantum chemistry calculations [3]. Other than the most computationally expensive model chemistries such as G1-G3 and CBS, the reaction energy errors of most computational chemistry model chemistries are well above 10 kJ mol−1.
For oxidation of the elements into binary compounds, an average error of ~4% or 33 kJ/mol-O2 is typical.[^9] For conventional ternary oxide formation from the elements, we have found a mean relative absolute error of about 2%. [4]
Sources of error
The largest contribution to the error comes from the inability of the GGA to fully describe electronic exchange and correlation effects. In addition, there is some error associated with neglecting zero-point effects and with comparing 0K, 0atm computations with room-temperature enthalpy experiments. The latter effect was estimated to contribute less than 0.03 eV/atom by Lany. [5] The stability of antiferromagnetic compounds may be underestimated, as the majority of our calculations are performed ferromagnetically only. The effect of magnetism may be small (under 10 meV/atom) or large (100 meV/atom or greater), depending on the compound. For compounds with heavy elements, relativistic effects may lead to greater-than-expected errors.
GGA errors on reaction energies between chemically similar compounds
We recently conducted a more in-depth study comparing GGA (+U) reaction energies of ternary oxides from binary oxides on 135 compounds. [6]
The main conclusions are:
The error in reaction energies for the binary oxide to ternary oxides reaction energies are an order of magnitude lower than for the more often reported formation energies from the element. An error intrinsic to GGA (+U) is estimated to follow a normal distribution centered in zero (no systematic underestimation or overestimation) and with a standard deviation around 24 meV/at.
When looking at phase stability (and for instance assessing if a phase is stable or not), the relevant reaction energies are most of the time not the formation energies from the elements but reaction energies from chemically similar compounds (e.g., two oxides forming a third oxide). Large cancellation of errors explain this observation.
The +U is necessary for accurate description of the energetics even when reactions do not involve change in formal oxidation states
Citations
To cite the calculation methodology, please reference the following works:
A. Jain, G. Hautier, C. Moore, S.P. Ong, C.C. Fischer, T. Mueller, K.A. Persson, G. Ceder., A High-Throughput Infrastructure for Density Functional Theory Calculations, Computational Materials Science, vol. 50, 2011, pp. 2295-2310. DOI:10.1016/j.commatsci.2011.02.023
A. Jain, G. Hautier, S.P. Ong, C. Moore, C.C. Fischer, K.A. Persson, G. Ceder, Accurate Formation Enthalpies by Mixing GGA and GGA+U calculations, Physical Review B, vol. 84, 2011, p. 045115. DOI:10.1103/PhysRevB.84.045115
References
[1]: Wang, A., Kingsbury, R.S., Horton, M., Jain, A., Ong, S.P., Dwaraknath, S., Persson, K. A framework for quantifying uncertainty in DFT energy corrections. Scientific Reports 11 (2021), 15496. DOI: 10.1038/s41598-021-94550-5
[2]: A. Jain, G. Hautier, S.P. Ong, C. Moore, C.C. Fischer, K.A. Persson, G. Ceder, Formation Enthalpies by Mixing GGA and GGA+U calculations, Physical Review B, vol. 84 (2011), 045115.
[3]: J.B. Foresman, A.E. Frisch, Exploring Chemistry With Electronic Structure Methods: A Guide to Using Gaussian, Gaussian. (1996).
[4]: A. Jain, S.-a Seyed-Reihani, C.C. Fischer, D.J. Couling, G. Ceder, W.H. Green, Ab initio screening of metal sorbents for elemental mercury capture in syngas streams, Chemical Engineering Science. 65 (2010) 3025-3033.
[5]: S. Lany, Semiconductor thermochemistry in density functional calculations, Physical Review B. 78 (2008) 1-8.
[6]: G. Hautier, S.P. Ong, A. Jain, C. J. Moore, G. Ceder, Accuracy of density functional theory in predicting formation energies of ternary oxides from binary oxides and its implication on phase stability, Physical Review B, 85 (2012), 155208
ν∗=VoV, where Vo is the volume at zero pressure.
Eo∗∗=E(ν=1)
Citation
To cite the EOS data in the Materials Project, please reference the following work:
Latimer, K., Dwaraknath, S., Mathew, K., Winston, D., Persson, K. A. Evaluation of thermodynamic equations of state across chemistry and structure in the materials project. NPJ Computational Materials.4, 1, 2057-3960 (2018). DOI:10.1038/s41524-018-0091-x
Authors
Katherine Latimer
Shyam Dwaraknath
Donny Winston
References
[1]: Birch, F. Finite elastic strain of cubic crystals. Physical Review.71, 11, 809–824 (1947).
[2]: Roy, B. and Roy, S. B. Applicability of isothermal three-parameter equations of state of solids: A reappraisal. Journal of Physics: Condensed Matter.17, 39, 6193–6216 (2005).
[3]: Murnaghan, F. D. The compressibility of media under extreme pressures. Proceedings of the National Academy of Sciences.30, 244–247 (1944).
[4]: Pack, D., Evans, W., James, H. The Propagation of Shock Waves in Steel and Lead. The Proceedings of the Physical Society.60, 1–8 (1948).
[5]: Poirier, J. P. and Tarantola, A. A logarithmic equation of state. Physics of the Earth and Planetary Interiors.109, 1-2, 1–8 (1998).
[6]: Dymond, J. H. and Malhotra, R. The Tait equation: 100 years on. International Journal of Thermophysics.9, 6, 941–951 (1988).
[7]: Vinet, P., Ferrante, J., Rose, J. H., Smith, J. R. Compressibility of solids. Journal of Geophysical Research.92, 9319–9325 (1987).
Convergence
Plane-wave energy cutoff and k-point density settings were selected such that formation energies converged within approximately 1 meV/atom for a benchmark set of 21 materials and were selected to be conservatively high [2]:
Formula
Spacegroup
Materials Project ID
AlN
P63mc
mp-661
Al2O3
R3c
mp-1143
References
[1] P. Wisesa, K. A. McGill, and T. Mueller, Efficient generation of generalized Monkhorst-Pack grids through the use of informatics, Phys. Rev. B 93, 1 (2016).
[2] R. Kingsbury, A. S. Gupta, C. J. Bartel, J. M. Munro, S. Dwaraknath, M. Horton, and K. A. Persson Phys. Rev. Materials 6, 013801 (2022)
A description of the methodology for constructing and interpreting compositional phase diagrams from the Materials Project (MP) website and API.
Introduction
A phase diagram is a calculation of the thermodynamic phase equilibria of multicomponent systems. It is an important tool in materials science for revealing 1) thermodynamic stability of compounds, 2) predicted equilibrium chemical reactions, and 3) processing conditions for synthesizing materials. However, the experimental determination of a phase diagram is an extremely time-consuming process, requiring careful synthesis and characterization of all phases in a chemical system.
Computational modeling tools, such as the density functional theory (DFT) methods used by the Materials Project, can accelerate compositional phase diagram construction significantly. By calculating the energies of all known compounds in a given chemical system (e.g. the lithium/iron/oxygen chemical system, Li-Fe-O), we can determine the phase diagram for that system at a temperature of K and pressure of atm. Furthermore, for systems comprised of predominantly solid phases open with respect to a gaseous element, approximations can be made as to the finite temperature and pressure phase diagrams.
In this section, we will describe the theory/methodology behind the calculation of compositional phase diagrams.
Methodology
This section will discuss how to construct phase diagrams from DFT-calculated energies. This is exact process done by the Materials Project (MP) for computing formation energies, thermodynamic stability, and phase diagrams. This methodology has been implemented in Python within the pymatgen package. Please see for brief examples of how to build phase diagrams on your own.
Calculating formation energy
The formation energy, , is the energy change upon reacting to form a phase of interest from its constituent components. The components typically used are the constituent elements. For a phase composed of components indexed by , the formation energy can be calculated as follows:
where is the total energy of the phase of interest, is the total number of moles of component , and is the total energy of component . Note that is often referred to as the chemical potential of the component, however, this is only rigorously true when working with Gibbs free energies, .
Example:
For barium titanate, BaTiO, the formation energy would be calculated as:
Typically, formation energies are normalized on a per-atom basis by dividing by the number of atoms in 1 mole of formula. For example, for BaTiO, the normalized per-atom formation energy would be calculated by dividing the above by 5 atoms.
Constructing the compositional phase diagram
The convex hull approach
To construct a phase diagram, one needs to compare the relative thermodynamic stability of phases belonging to the system using an appropriate free energy model. For an isothermal, isobaric, closed system, the relevant thermodynamic potential is the Gibbs free energy, , which can be expressed as a Legendre transform of the enthalpy, , and internal energy, , as follows:
where is the temperature of the system, is the entropy of the system, is the pressure of the system, is the volume of the system, and is the number of atoms of species in the system.
For systems comprising primarily of condensed phases, the term can be neglected and at 0K, the expression for simplifies to just . Normalizing with respect to the total number of particles in the system, we obtain . By taking the convex hull of for all phases belonging to the M-component system and projecting the stable nodes into the - dimension composition space, one can obtain the 0 K phase diagram for the closed system at constant pressure. The convex hull of a set of points is the smallest convex set containing the points. For instance, to construct a 0 K, closed system phase diagram, the convex hull is taken on the set of points in space with being related to the other composition variables by .
Evaluating thermodynamic stability
Figure 2 is an example of a calculated binary A-X phase diagram at 0 K and 0 atm. Binary phase diagrams show the complete convex hull for the system, where the y-axis is the formation energy per atom and the x-axis is the composition.
The blue lines show the convex hull construction, which connects stable phases (circles). Unstable phases will always appear above the convex hull line (squares); one measure of the thermodynamic stability of an arbitrary compound is its distance from the convex hull line (), which predicts the decomposition energy of that phase into the most stable phases.
Accuracy of Calculated Phase Diagrams
In general, we can expect that compositional phase diagrams comprising of predominantly solid phases to be reproduced fairly well by our calculations. However, it should be noted that there are inherent limitations in accuracy in the DFT calculated energies. Furthermore, our calculated phase diagrams are at 0 K and 0 atm, and differences with non-zero temperature phase diagrams are to be expected.
For grand potential phase diagrams, further approximations are made as to the entropic contributions . They are therefore expected to be less accurate, but nonetheless provide useful insights on general trends.
Code (pymatgen)
While the Materials Project website has a phase diagram app (), and PhaseDiagram objects can also be obtained directly from the API (), two code snippets are provided below that show how to use the API and pymatgen to construct and plot your own phase diagrams with Python.
GGA/GGA+U
Constructing mixed GGA/GGA+U phase diagrams can be done directly with the corrected ComputedStructureEntry objects from the API.
GGA/GGA+U/R2SCAN
Constructing a mixed GGA/GGA+U/R2SCAN phase diagram requires corrections to be reapplied locally. This is because the corrected ComputedStructureEntry object obtained from the thermodynamic data endpoint of the API for a given material is from its home chemical system phase diagram (i.e. Si-O for SiO2, or Li-Fe-O for Li2FeO3).
Unlike the previous GGA/GGA+U only mixing scheme, the updated scheme does not guarantee the same correction to an entry in phase diagrams of different chemical systems. In other words, the energy correction applied to the entry for silicon (mp-149) in the Si-O phase diagram is not guaranteed to be the same for the one in the Si-O-P phase diagram.
For more details on the correction scheme and its logic, see the section or the original publication .
Citations
Methodology (I)
Methodology (II)
.
References
[1] Bartel, C.J. Review of computational approaches to predict the thermodynamic stability of inorganic solids. J Mater Sci57, 10475–10498 (2022).
[2] V. Raghavan, Fe-Li-O Phase Diagram, ASM Alloy Phase Diagrams Center, P. Villars, editor-in-chief; H. Okamoto and K. Cenzual, section editors; , ASM International, Materials Park, OH, 2006.
[3]:
[4] Kingsbury, R.S., Rosen, A.S., Gupta, A.S. et al. A flexible and scalable scheme for mixing computed formation energies from different levels of theory. npj Comput Mater 8, 195 (2022).
Aqueous Stability (Pourbaix)
How aqueous stability (Pourbaix) diagrams are calculated and plotted on the Materials Project (MP) website.
If using Pourbaix functionality for scientific research, please make sure to consult the original peer-reviewed publication and the section below.
Introduction
Version History
Changes made to the QMOF Database: https://doi.org/10.6084/m9.figshare.13147324
Changelog
: New single-point calculations at the HLE17, HSE06* (i.e. 10% HF ex.), and HSE06 (25% HF ex.) levels of theory. 12/09/21.
v12: Several MOFs taken from the Genomic MOF Database with over/underbonded atoms were removed, as the original authors of the Genomic MOF Database uploaded a fairly large fraction of structures with missing H atoms. Supplemental results from new non-self-consistent (NSCF) calculations with a higher k-point density are now provided (note: this was reverted in v13, as it was discovered that LMAXMIX was not set high enough, effecting the NSCF results for a subset of structures). Removed raw VASP files from Figshare to instead host them on NOMAD. Gave each MOF a unique hash-based identifier, which will match the identifiers on the forthcoming Materials Project MOF Explorer app. 09/14/21.
v11: Same changes as in v12, but the bandgaps.csv file was not made backwards-compatible here. 09/13/21.
v10: Removed irrelevant data from the JSON, reducing the filesize. 09/01/21.
v9: Added ~3000 new DFT-optimized MOFs from the CoRE MOF Database (based on the clean subset identified by Chen and Manz), the Genomic MOF Database, and the CSD MOF Subset. Deprecated 13 structures. Added spacegroup info. Added "synthesized?" flag. Added missing PLDs and LCDs. Fixed 186 structures that had EDIFF = 1e-4 instead of EDIFF = 1e-6 in the INCAR. Removed structures that were duplicates according to Pymatgen's StructureMatcher to avoid confusion. The user no longer needs to run the StructureMatcher as a result. 09/01/21.
v8: Added 1243 new DFT-optimized MOFs. 623 were taken from the Boyd & Woo dataset, 485 were taken directly from the 2019 CoRE MOF FSR Database, 92 were Cu triangle MOFs taken from ToBaCCo, and 44 were Hf MOFs obtained by exchanging the Zr metals of ToBaCCo MOFs by Anderson and coworkers. For the CoRE MOFs, only those found in this pre-curated list were included to maximize structural fidelity. For the hypothetical MOFs, some new ones were introduced using the Boyd & Woo structures as a starting point (e.g. by exchanging metal cations). 3 MOFs were deprecated. Added MOFids, DOIs, spin-dependent CBM/VBM, and initial CIFs for the hypothetical MOFs. 07/12/21.
v7: Deprecated 12 MOFs. Added more properties to JSON file and made it easier to parse. 06/08/21.
v6: Added 2620 DFT-optimized MOFs. 1217 were taken from the CSD using the usual protocol. 1188 were hypothetical MOFs obtained from the Boyd & Woo dataset. 148 were hypothetical MOF-74 and MOF-5 analogues obtained from Haranczyk's nanoporousmaterials.org. 48 were hypothetical Zr MOFs made with ToBaCCo and obtained from Anderson and coworkers. 19 were experimental pyrene MOFs from Smit and coworkers. The maximum number of atoms per unit cell was raised to 500. 5/7/2021.
v5: Release corresponding to the published Matter paper. No changes to the database compared to v3. Fixes a bug in get_subset_data.py that did not correctly write out the updated .json file. 2/12/21.
v4: Includes a few minor typo fixes and better .xlsx reader. 1/12/21.
v3: Added CM5 partial charges for every structure and 3000+ Bader charges (and spin densities). Patched some minor bugfixes with the unrelaxed properties for a few MOF structures, deprecated a few structures, and flagged more duplicates. Continued restructuring of main QMOF database for increased useability. 12/23/20.
v2: ~1500 new structures with pore-limiting diameter greater than 2.4 Å, computed using Zeo++ prior to structure relaxation, were added to the QMOF database along with their DFT-computed properties. The cap on the maximum number of atoms per primitive cell was raised from 150 to 300. 12/05/20.
v1: Initial release corresponding to the QMOF database pre-print. 10/28/20.
A Pourbaix diagram, also frequently called a potential-pH diagram, or E-pH diagram, is a representation of aqueous phase electrochemical equilibria. It is a two-dimensional representation of a three-dimensional free energy-pH-potential diagram. In other words, it shows water-stable phases as a function of pH and potential, where, potential is defined with respect to the standard hydrogen electrode.
Experimentally determining Pourbaix Diagrams is painstaking work, as we need not only the free energy of aqueous ions, but also that of all solid phases that a system can exist in. The Materials Project offers a very convenient and powerful database of materials properties which has been used to generate Pourbaix diagrams in a high-throughput manner.
This manual outlines the usage of the Pourbaix App to calculate Pourbaix diagrams, and the thermodynamic formalism underlying the app.
Thermodynamic Formalism of Pourbaix Diagrams
To calculate a Pourbaix diagram, free energies of the solid phases, and of the aqueous ions are required. Calculating free energies of ions is tricky, and time-consuming. To overcome this problem, a methodology utilizing experimentally measured free energies of aqueous ions and the calculated DFT energies for solid phases available in the Materials Project was developed.[1] Note that the correction scheme described below is applied over and above any compatibilities/corrections which are applied to the species.
Referencing Energies of Aqueous Ions
Briefly, for each ion, a reference solid is chosen, and the correction term is calculated for the ion as the energy difference between the experimental and the DFT calculated energies of the reference solid. The basic idea behind this scheme is that, if we have a reference energy for an aqueous ion which reproduces the correct dissolution for one solid, then accurate DFT solid-solid energy differences ensure that all other solids dissolve accurately with respect to that ion. The better the solid is represented by DFT, the more transferable the reference aqueous energy becomes. We therefore prefer to choose simple chemical systems (primarily binaries with an uncomplicated electronic structure) as representative solids.
For an aqueous ion i at standard state conditions (e.g., room temperature, atmospheric pressure, and 10^−6 M concentration) using a representative solid s, we define the chemical potential as:
Figure 1: Schematic to reference experimental aqueous ion energies to DFT data
Figure 1 shows this schematically.
Correction for Water
In an aqueous environment, many chemical and electrochemical reactions are enabled by the breakdown, formation, or incorporation of water molecules. It is therefore important that the free energy of formation of water is captured accurately. This is known accurately from experiments as −2.46 eV. So, at standard state, the free energy of formation of water is set as follows:
Correction for Elemental Gaseous, and Liquid States, except H2
For all gaseous elements, the experimentally determined entropic contribution at 298 K is added to the DFT/corrected energy of the element as follows:
This is implemented for the following elements: O2, F2, Cl2, Br2, Hg
Correction for H2
In an aqueous environment, O2 and H2 in their gaseous states are in equilibrium with water through the reaction
Hence, the hydrogen energy is corrected such that the experimental free energy of formation of H2O is reproduced.
Electrochemical Stability of Metastable Materials
In principle, Pourbaix diagrams account for materials only at thermodynamic equilibrium, providing no insight into the electrochemical stability of metastable materials which find practical applications in many commercial applications. However, one can compute the Gibbs free energy difference for an arbitrary material with respect to the Pourbaix stable domains as a function of pH and E, providing an electrochemical (in)stability map for this material. For detailed information on the formalism and its applications see reference 2.[2]
Using the Computational Pourbaix App
The Pourbaix app is capable of plotting elemental, and multi-element Pourbaix diagrams. To construct an Elemental Pourbaix diagram, enter the element of choice, and click on the Generate button.
To generate a multi-elemental Pourbaix diagram, choose the desired multiple elements from the periodic table, and click the Generate button. Note that oxygen and hydrogen are included by default, since these elements are always "open" in a Pourbaix diagram.
For multi-elemental diagrams, sliders are provided to specify a composition of the elements. Note that for each composition, a new Pourbaix diagram is generated from scratch.
To generate electrochemical stability maps of a specific material go to the material's details page and click on the "Aqueous Stability (Pourbaix)" tab which can be found in the "Generate Phase Diagram" tab. A new tab will open which will show the electrochemical stability map superimposed on a Pourbaix diagram. The ratio of elements used to generate the Pourbaix diagram are same as that of the material in consideration. Electrochemical stability maps are available for materials with up to three non H and O elements. For materials with more than three elements, one can use pymatgen to generate the maps programmatically.
Interfacing With the Data
To ensure a clutter-free diagram, the domains on the Pourbaix diagram are not labeled by default. To view labels on the plot, select the "Domain Labels" check box. Each domain has a mouse-over point located at the center of the domain. Mousing over these points displays the entries corresponding to the domain. Domains which contain a solid in solution are shaded. This helps identify passivation regions, especially in multi-elemental systems, were identifying passivation regions is tricky. Zooming into domains is quite simple! Just drag a selection window using your mouse. To reset zoom, click on the reset zoom button which appears on the upper right corner. Data tables are shown to the right of the Pourbaix diagram. Mousing over rows in the "Stable" column of the table highlights the corresponding entries in the Pourbaix diagram. Some stable entries may not get highlighted. This is because the domains corresponding to these entries lies outside the standard limits of the Pourbaix diagram. The "Unstable" column in the table lists the unstable entries, and their corresponding energies above hull. Links in the data tables lead to more information about the corresponding entries. Unlinked entries are ions. Mousing over the book icon next to the ions shows the reference for the free energy of formation of the aqueous ion.
Demonstration of the Pourbaix App
This section briefly demonstrates an elemental Pourbaix diagram, a multi-elemental Pourbaix diagram, and an electrochemical stability map.
Elemental Pourbaix Diagram
Figure 2 shows the elemental Pourbaix diagram for Fe. The default concentration of ions is 10−8 M, but can be varied using the "Concentration" slider above the diagram.
Figure 2: Pourbaix diagram for elemental Fe.
The two orange lines are the hydrogen reduction line, and the line denoting water oxidation to O2. These are clearly labeled in Figure 3. These lines show the stability region of H2O. For example, water is unstable below the H2 line, and so, hydrogen gas evolves at the cathode at conditions below this line. Similarly, above the O2 line, oxygen gas is evolved at the anode.
Figure 3: Orange lines are the water oxidation and reduction lines in the Pourbaix diagram.
Multi-elemental Pourbaix diagram
The stability of multiple-elements in aqueous environments is predicted using multi-elemental Pourbaix diagrams like the one shown in Figure 4. The composition slider bar can be seen above the plot. Here, the small white bar separating the two colors can be clicked on and dragged to change the ratio of Fe to Cr. This may, or may not have any effect on the Pourbaix diagram. More information about how multi-elemental Pourbaix diagrams vary as a function of composition can be found elsewhere.[3]
Figure 4: Multi-elemental Fe-Cr Pourbaix diagram at a composition of 18% Cr, 82% Fe.
For an n-element diagram, there are n coexisting phases. Note that these can be any mixture of solid, and aqueous phases. So, for the two-element diagram shown in Figure 4, there are two coexisting phases in each domain. As mentioned above, domains shaded blue indicate purely solid domains. For multi-elemental diagrams, shaded domains indicate those in which purely solid phases coexist.
Electrochemical Stability Map
Figure 5 shows the Gibbs free energy of Fe2O3, as a scatter plot, superimposed over the Pourbaix diagram of Fe. For a material with more than one non H and O elements, the ratio of these elements is fixed to the ratio of elements of the material but the concentration of ions can be varied similar to the single- and multi-element Pourbaix diagrams.
Figure 5: Electrochemical stability map of Fe2O3 (mp-24972).
A color bar is shown above the electrochemical stability maps. Note that Gibbs free energies larger than 1 eV/atom are not marked in the map. Stable and unstable phase energies can be found in the table to the right of the electrochemical stability map.
Literature References for Ions
The free energies of ions in the aqueous phase have been taken from standard references/recent publications. The acronyms which show up on the tool-tips associated with the aqueous ions, and their corresponding references are as follows.
NBS Tables: NBS Thermodynamic tables.4 M. Pourbaix (1974): Atlas of Electrochemical Equilibria in Aqueous Solutions. 5 Barin Knacke Kubaschewski: Thermochemical Properties of Inorganic Substances 6 Barner and Scheuerman (1978): Handbook of thermochemical data for compounds and aqueous species 7 Beverskog and Puigdomenech (1997): Beverskog and Puigdomenech, Corr. Sci. (1997) 8
Authors
Sai Jayaraman
Arunima K. Singh
Rebecca Stern
Eric Sivonxay
Citations
K. A. Persson, B. Waldwick, P. Lazic, and G. Ceder, Phys. Rev. B, 85, 235438 (2012) ↩
A. K. Singh, L. Zhou, A. Shinde, S. K. Suram, J. H. Montoya, D. Winston, J. M. Gregoire, K. A. Persson, Chem. Mater. 29, 10159 (2017) ↩
Pourbaix Diagrams for Multielement Systems, Thompson, W. T., Kaye, M. H., Bale, C. W. and Pelton, A. D. (2011), in Uhlig's Corrosion Handbook, Third Edition (ed R. W. Revie), John Wiley & Sons, Inc., Hoboken, NJ, USA. ↩
NBS Technical Note 270-1 to 270-8. D. D. Wagman et. al, U. S. Department of Commerce (1973)
Atlas of Electrochemical Equilibria in Aqueous Solutions, M. Pourbaix, NACE (1974)
Thermochemical Properties of Inorganic Substances, I. Barin, O. Knacke, and O. Kubaschewski, Springer-Verlag, Berlin (1977)
Handbook of thermochemical data for compounds and aqueous species, H. E. Barner, and R. V. Scheuerman, Wiley, New York, 1978
Revised Pourbaix diagrams for Ni at 25-300^oC, B. Beverskog and I. Puigdomenech, Corr. Sci., 39, 969-980 (1997)
Details on Hubbard U corrections used by the Materials Project
Hubbard U Values
It is well-known that first principles calculations within the local density approximation (LDA) or generalized gradient approximation (GGA) lead to considerable error in calculated redox reaction energies of many transition metal compounds. This error arises from the self-interaction error in LDA and GGA, which is not canceled out in redox reactions where an electron is transferred between significantly different environments, such as between a metal and a transition metal or between a transition metal and oxygen or fluorine. Extensive discussion of this issue can be found in the following works. [1-4]
In the Materials Project, we have calibrated U values for many transition metals of interest using the approach outlined in Wang et al.'s work . At the present moment, values have only been calibrated for transition metal oxide systems. values were calibrated for the following elements: , , , , , , and . The choice of systems to which we apply was largely determined by our experience and by systematic benchmarking. It is very likely that we will expand calibration of values to more chemical systems in the future.
In the Materials Project, for an oxide or fluoride material with a transition element listed previously, with the VASP input settings constructed according to the logic defined in .
Note that for fluorides, the value gets set to the one calibrated from the oxide system, although in principle our architecture allows different values to be set for oxides and fluorides respectively.
Calibration and Values
The values were obtained by fitting to experimental binary formation enthalpies as described in Wang et al.'s work. This method is simple yet accurately reproduces phase stabilities. A least squares method of obtaining the correct value was used, as follows:
For each non-overlapping formation energy reaction considered, we find the region where the formation energy error passes zero. For the system, this includes the following:
The full list of U values used is described in the table below. For oxides and fluorides containing any of the elements, only GGA+U calculations are performed.
Element
System
Fitting Reaction
Redox Couple
Calibrated U (eV)
Comments
Caveats
The U values are calibrated for phase stability analyses, and should be used with care if applied to obtain other properties such as band structures. Also, the U values depend on the pseudopotential used. Further, typically, U values should be site specific, however in our approach, U values were applied to all sites with an element listed above, and only to the d-orbitals. A discussion of the pseudopotentials used in the Materials Project can be found .
References
[1]: F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder. First-principles prediction of redox potentials in transition-metal compounds with LDA+U. Physical Review B, 2004, 70, 235121.
[2]: M. Cococcioni, S. de Gironcoli, Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Physical Review B, 2005, 71, 035105.
[3]: L. Wang, T. Maxisch, & G. Ceder. Oxidation energies of transition metal oxides within the GGA+U framework. Physical Review B. 2006, 73, 195107,
[4]: A. Jain, G. Hautier, S. P. Ong, C. Moore, C. Fischer, K. A. Persson, & G. Ceder. Formation enthalpies by mixing GGA and GGA + U calculations. Physical Review B, 2011, 84(4), 045115.
[5]: M. Wang, A. Navrotsky Enthalpy of formation of LiNiO2, LiCoO2 and their solid solution, LiNi1-xCoxO2, Solid State Ionics, vol. 166, no. 1-2, pp. 167-173, Jan. 2004.
Related Materials
How related materials are identified on the Materials Project (MP) website.
Introduction
The similarity between two structures i and j is assessed on the basis of local coordination information from all sites in the two structures. The four basic steps involved are:
from mp_api.client import MPRester
from pymatgen.analysis.phase_diagram import PhaseDiagram, PDPlotter
with MPRester("your_api_key") as mpr:
# Obtain only corrected GGA and GGA+U ComputedStructureEntry objects
entries = mpr.get_entries_in_chemsys(elements=["Li", "Fe", "O"],
additional_criteria={"thermo_types": ["GGA_GGA+U"]})
# Construct phase diagram
pd = PhaseDiagram(entries)
# Plot phase diagram
PDPlotter(pd).get_plot()
from mp_api.client import MPRester
from pymatgen.analysis.phase_diagram import PhaseDiagram, PDPlotter
from pymatgen.entries.mixing_scheme import MaterialsProjectDFTMixingScheme
with MPRester("your_api_key") as mpr:
# Obtain GGA, GGA+U, and r2SCAN ComputedStructureEntry objects
entries = mpr.get_entries_in_chemsys(elements=["Li", "Fe", "O"],
additional_criteria={"thermo_types": ["GGA_GGA+U", "R2SCAN"]})
# Apply corrections locally with the mixing scheme
scheme = MaterialsProjectDFTMixingScheme()
corrected_entries = scheme.process_entries(entries)
# Construct phase diagram
pd = PhaseDiagram(corrected_entries)
# Plot phase diagram
PDPlotter(pd).get_plot()
Find near(est) neighbors of all sites in both structures.
Evaluate each coordination pattern via coordination descriptors observed at each site to define site fingerprints.
Compute statistics of the descriptor values across all sites in a structure to define structure fingerprints.
Use structure fingerprints to rate the (dis)similarity between the two (vectors representing the two) structures.
Near-neighbor finding
We use a novel method called to find near(est) neighbors in periodic structures. While the method will be introduced shortly , it is already available through the python package . A benchmarking framework has been developed to evaluate CrystallNN and compare it to other near-neighbor finding algorithms .
Site Fingerprints
The second step of the structure similarity calculation is the computation of a crystal site fingerprint, , for each site in the two structures. The fingerprint is a 61-dimensional vector in which each element carries information about the local coordination environment computed with the site module of the python package . For example, the first two elements "wt " and "single bond " provide estimates of the likelihood (or weight) of how much the given site should be considered 1-fold coordinated (i.e., w). The third element "wt " provides a 2-fold coordination likelihood, whereas the fourth element "L-shaped " holds the resemblance similarity to an L-shaped coordination geometry (also called local structure order parameter) given that we find a coordination configuration with 2 atoms (). The local structure order parameters can assume values between 0, meaning that the observed local environment has no resemblance with the target motif to which it is compared, and 1, which stands for perfect motif match. The remaining elements are: "water-like ", "bent 120 degrees ", "bent 150 degrees ", "linear ", "wt ", "trigonal planar ", "trigonal non-coplanar ", "T-shaped ", "wt ", "square co-planar ", "tetrahedral ", "rectangular see-saw-like ", "see-saw-like ", "trigonal pyramidal ", "wt
Structure Fingerprints
The fingerprints from sites in a given structure are subsequently statistically processed to yield the minimum, maximum, mean, and standard deviation of each coordination information element," The resultant ordered vector defines a structure fingerprint, $v^{struct}$:
Structure Distance/Dissimilarity
Finally, structure similarity is determined by the distance, d, between two structure fingerprints and:
A small distance value indicates high similarity between two structures, whereas a large distance (>1) suggests that the structures are very dissimilar," The spinel example below gives an approximate threshold up to which distance you can still consider two structures to be similar (0.9)," Anything beyond 0.9 is most certainly not the same structure prototype.
Examples
Diamond () vs. () d = 0
Diamond () vs. rocksalt () d = 3.5724
Diamond (
Below is a python code snippet that allows you to quickly reproduce above results," You will need to install and for this to work," Both are easily accessible via the .
StructureMatcher
Another tool that is used to group materials is the . There are multiple comparators (for example: , , etc.) that can be used to determine how to make comparisons between structures when determining their similarity.
References
[1]: N. E. R. Zimmermann, D. Winston, K. A. Persson, A. Jain, in preparation (2018)
[2]: 10.3389/fmats.2017.00034
[3]: H. Pan, J. Dagdelen, N. E. R. Zimmermann, A. Jain, in preparation (2018)
[4]: Pan, H., Ganose, A. M., Horton, M., Aykol, M., Persson, K. A., Zimmermann, N. E., & Jain, A. (2021). Benchmarking coordination number prediction algorithms on inorganic crystal structures. Inorganic chemistry, 60(3), 1590-1603.
Authors
Nils Zimmermann, Donny Winston, Handong Ling, Oxana Andriuc
For solid state materials, the VASP 5.4 PBE projector-augmented wave (PAW) pseudopotentials were used to carry out the DFT calculations. In general, the VASP-recommended PAW PBE potentials were adopted except for Li, Eu_3, Yb_3, and W_sv. The full list of pseudopotentials is shown below:
Element
VASP 5.4 Pseudopotentials
Elastic Constants
How elastic constants are calculated on the Materials Project (MP) website.
Introduction
Elasticity describes a material's ability to resist deformations (i.e. size and shape) when subjected to external forces. This can be thought about in two, complementary ways:
how much force is required to deform (stretch or compress) a material by a certain amount;
Ag
PAW_PBE Ag 02Apr2005
Al
PAW_PBE Al 04Jan2001
As
PAW_PBE As 22Sep2009
Au
PAW_PBE Au 04Oct2007
B
PAW_PBE B 06Sep2000
Ba
PAW_PBE Ba_sv 06Sep2000
Be
PAW_PBE Be 06Sep2000
Bi
PAW_PBE Bi_d 06Sep2000
Br
PAW_PBE Br 06Sep2000
C
PAW_PBE C 08Apr2002
Ca
PAW_PBE Ca_sv 06Sep2000
Cd
PAW_PBE Cd 06Sep2000
Ce
PAW_PBE Ce 23Dec2003
Cl
PAW_PBE Cl 06Sep2000
Co
PAW_PBE Co 02Aug2007
Cr
PAW_PBE Cr_pv 02Aug2007
Cs
PAW_PBE Cs_sv 08Apr2002
Cu
PAW_PBE Cu 22Jun2005
Dy
PAW_PBE Dy_3 06Sep2000
Er
PAW_PBE Er_3 06Sep2000
Eu
PAW_PBE Eu_3 20Oct2008
F
PAW_PBE F 08Apr2002
Fe
PAW_PBE Fe 06Sep2000
Ga
PAW_PBE Ga_d 06Jul2010
Gd
PAW_PBE Gd_3 06Sep2000
Ge
PAW_PBE Ge_d 03Jul2007
H
PAW_PBE H 15Jun2001
Hf
PAW_PBE Hf_pv 06Sep2000
Hg
PAW_PBE Hg 06Sep2000
Ho
PAW_PBE Ho_3 06Sep2000
I
PAW_PBE I 08Apr2002
In
PAW_PBE In_d 06Sep2000
Ir
PAW_PBE Ir 06Sep2000
K
PAW_PBE K_sv 06Sep2000
La
PAW_PBE La 06Sep2000
Li
PAW_PBE Li 17Jan2003
Lu
PAW_PBE Lu_3 06Sep2000
Mg
PAW_PBE Mg 13Apr2007
Mn
PAW_PBE Mn_pv 02Aug2007
Mo
PAW_PBE Mo_sv 02Feb2006
N
PAW_PBE N 08Apr2002
Na
PAW_PBE Na_pv 19Sep2006
Nb
PAW_PBE Nb_sv 25May2007
Nd
PAW_PBE Nd_3 06Sep2000
Ni
PAW_PBE Ni 02Aug2007
Np
PAW_PBE Np 06Sep2000
O
PAW_PBE O 08Apr2002
P
PAW_PBE P 06Sep2000
Pb
PAW_PBE Pb_d 06Sep2000
Pd
PAW_PBE Pd 04Jan2005
Pr
PAW_PBE Pr_3 07Sep2000
Pt
PAW_PBE Pt 04Feb2005
Pu
PAW_PBE Pu 06Sep2000
Rb
PAW_PBE Rb_sv 06Sep2000
Re
PAW_PBE Re 17Jan2003
Rh
PAW_PBE Rh_pv 25Jan2005
Ru
PAW_PBE Ru_pv 28Jan2005
S
PAW_PBE S 06Sep2000
Sb
PAW_PBE Sb 06Sep2000
Sc
PAW_PBE Sc_sv 07Sep2000
Se
PAW_PBE Se 06Sep2000
Si
PAW_PBE Si 05Jan2001
Sm
PAW_PBE Sm_3 07Sep2000
Sn
PAW_PBE Sn_d 06Sep2000
Sr
PAW_PBE Sr_sv 07Sep2000
Tb
PAW_PBE Tb_3 06Sep2000
Tc
PAW_PBE Tc_pv 04Feb2005
Te
PAW_PBE Te 08Apr2002
Th
PAW_PBE Th 07Sep2000
Ti
PAW_PBE Ti_sv 26Sep2005
Tl
PAW_PBE Tl_d 06Sep2000
Tm
PAW_PBE Tm_3 20Jan2003
U
PAW_PBE U 06Sep2000
V
PAW_PBE V_sv 02Aug2007
W
PAW_PBE W_sv 04Sep2015
Y
PAW_PBE Y_sv 25May2007
Yb
PAW_PBE Yb_3 08Jul2013
Zn
PAW_PBE Zn 06Sep2000
Zr
PAW_PBE Zr_sv 04Jan2005
how much a material will deform (stretch or compress) when a certain amount of external forces is applied to that material.
Elasticity is considered a reversible process. When the force is removed, the material returns to its original size and shape. This is only true up to a point: if a material is deformed too much, then it will be permanently changed.
For small deformations, most elastic materials exhibit linear elasticity and can be described by a linear relation between the stress and strain. These relationships are quantified with elastic constants like the elasticity tensorand its inverse quantity, the compliance tensor, as part of the theory of linear elasticity. These tensors can be used to calculate numbers such as the bulk modulus, shear modulus, Young's modulus, and Poisson's ratio, which are especially useful to describe the elastic behavior of isotropic materials.
The Materials Project predicts elastic constants for over ten thousand materials. These are available via the Materials Project website and for direct download via the Materials Project API.
Methodology
Overview
The elastic constants from the Materials Project (MP) are calculated from first-principles Density Functional Theory (DFT). For a material, the process is started by performing an accurate structural relaxation, to a state of approximately zero stress. Subsequently, the relaxed structure is strained by changing its lattice vectors (magnitude and angle) and the resulting stress tensor is calculated from DFT, while allowing for relaxation of the ionic degrees of freedom. Finally, constitutive relations from linear elasticity, relating stress and strain, are employed to fit the full elastic tensor. From this, aggregate properties such as Voigt, Reuss, and Hill bounds on the bulk and shear moduli are derived. Multiple consistency checks are performed on all the calculated data to ensure its reliability and accuracy. For example, the 6×6 Voigt elastic matrix should be positive definite to ensure mechanical stability of a material.
Voigt notation
Formally, the elastic tensor, C^, is a forth-order tensor with 81 components (but only with 21 independent components):
where σ and ϵ are the second-order stress and strain tensors, respectively, and i,j,k,l are Cartesian indices, taking values x, y, and z. Both σ and ϵ symmetric tensor, and we can represent them in Voigt notation under the transformation xx↦1,yy↦2,zz↦3,yz↦4,xz↦5,xy↦6. For example, the strain transforms like ϵ1=ϵxx,ϵ2=ϵyy,ϵ3=ϵzz,ϵ4=ϵyz,ϵ5=ϵxz,ϵ6=ϵxy, and the elastic tensor transforms like Cxxxx↦C11,Cxxyy↦C12,.....Then the above linear elastic relationship can be expressed as
The elastic tensor in Voigt notation is a 6×6 symmetric matrix, indicating that the elastic tensor has 21 independent components.
Formalism
With the lattice vectors{a1,a2,a3} of the relaxed structure, a material is first deformed according to a^i=Fai,(i=1,2,3). The deformation gradient F is obtained by solving the equation for Green-Lagrange strain E, namely ϵ=E=21(FTF−I), where I is the identify matrix and the superscript denotes matrix transpose. Then he stress tensor, σ, is obtained from DFT calculation for the deformed structure with the new lattice vectors {a^1,a^2,a^3}. In the DFT calculation, the lattice vectors are fixed, but the ionic degree of freedoms are allowed to relax. Six strain states (listed below) are applied one by one to the initial relaxed structure so that only one independent deformation is considered each time. For each of the six strain states, 4 different default magnitudes strains are applied: δ∈{−0.01,−0.005,+0.005,+0.01}.This leads to a total of 24 deformed structures, for which the stress tensor, σ, is calculated. The obtained set of 24 stresses and strains are then used in a linear fitting to compute the elastic tensor. Note that conventional unit cells, obtained using pymatgen SpacegroupAnalyzer, are employed for all elastic constant calculations. In our experience, these cells typically yield more accurate and better converged elastic constants than primitive cells, at the cost of more computational time. We suspect this has to do with the fact that unit cells often exhibit higher symmetries and simpler Brillouin zones than primitive cells (an example is face centered cubic cells).
Different choices of lattice vectors with respect to a Cartesian coordinate system may lead to elastic tensors that look different from what might be expected. For example, for the hexagonal crystal system it is commonly stated that C11=C22. However, this is true under the conditions that lattice vectors a1anda2are both in the basal plane, whereas a3 is orthogonal to the basal plane. Hence, the elastic tensor can only be completely specified when the lattice vectors are expressed in a given coordinate system. To avoid confusion, we present the elastic tensor in two ways. First, the elastic tensor is presented for the exact choice of lattice vectors as presented on the Materials Project webpage. This is consistent with the cif-file of the "conventional standard" structure, which can also be downloaded from the Materials Project webpage. Elastic tensors can also be expressed in a standard format according to the IEEE standard. The standardized IEEE-format specifies the precise choice of lattice vectors in a coordinate system and thereby unambiguously defines the components of the elastic tensor [1]. In most cases, the elastic tensors in the POSCAR-format and the IEEE-format are identical. When the elastic tensor in POSCAR-format and IEEE-format are not identical however, they are related by a rotation, which can be obtained using the get_ieee_rotation method pymatgen.core.tensors.Tensor (including the elastic tensor).
Derived elastic properties
From the elastic tensor defined above, a number of aggregate and derived properties is calculated. These properties are all available on the Materials Project webpage and are shown in the below Table. We report Voigt, Reuss and Voigt-Reuss-Hill [2] bounds on the bulk and shear moduli for polycrystalline materials. Finally, the elastic anisotropy index [3] and isotropic Poisson ratio are reported.
Property
Unit
Description
Equation
Elastic tensor,
GPa
Tensor, describing elastic behavior, corresponding to IEEE orientation, symmetrized to crystal structure
see main text
DFT parameters
To obtain accurate elastic constants from DFT, a well-converged stress tensor is required. This typically means that more precise DFT-parameters have to be employed, compared to for example a simple total energy-calculation. Careful convergence testing and comparison to experimental results has led to a set of DFT-parameters that yield elastic constants, converged to within approximately 5% for over 95% of the systems. In choosing DFT-parameters for the calculations, we distinguish between metals and metallic compounds (metallics) on one hand and semiconductors and insulators (non-metallics) on the other hand. The most relevant DFT-parameters used in our HT-calculations are shown in Table 2. K-point density is expressed in per-reciprocal-atom (pra). The first-principles results presented in this work are performed using the projector augmented wave (PAW) method [3,4] as implemented in the Vienna Ab Initio Simulation Package (VASP) [5,6,7] . In all calculations, we employ the Perdew, Becke and Ernzerhof (PBE) [8] Generalized Gradient Approximation (GGA) for the exchange-correlation functional. As described in the literature, several filters are used to detect cases where the elastic tensor might not have been converged properly. For those cases, the calculation is repeated but now with more stringent DFT-convergence parameters. Hence, the numerical values in Table 2 are representative for our calculations, but in some cases more strict parameters have been used. The calculation details for each compound can be found on the Materials Project webpage.
Metallics
Non-metallics
Plane wave energy cut-off (eV)
700
700
Density of k-points (pra)
7,000
Visualization of the current elastic-property database, consisting of over 1,100 metals and inorganic compounds. This map shows the shear and bulk moduli, together with isotropic Poisson ratio and volume-per-atom. See the paper [Charting the complete elastic properties of inorganic crystalline compounds](http://www.nature.com/articles/sdata20159) for details.
Symmetrization
Tensor symmetrization and IEEE conversion procedures are implemented in pymatgen. Symmetrization occurs by finding all of the symmetry operations that correspond to a particular crystal symmetry, and taking the average over all transformed tensors with respect to these operations. If there are y symmetry operations are denoted Qij(x) then:
How to Cite
If you use any elastic constants predicted by the Materials Project in your work, the corresponding methods paper(s) should be cited. See the How to Cite page for more.
Thanks
Thanks to Maarten de Jong for the initial version of this page.
References
IEEE standard on piezoelectricity. ANSI/IEEE Std 176-1987 0-1 (1988).
Hill, R. The elastic behaviour of a crystalline aggregate. Proceedings of the Physical Society. Section A 65, 349 (1952).
Ranganathan, S. I. & Ostoja-Starzewski, M. Universal elastic anisotropy index. Physical Review Letters 101, 055504 (2008).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953{17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B59, 1758{1775 (1999).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558{561 (1993).
Kresse, G. & Furthmuller, J. Efficffient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169{11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Physical Review Letters 77, 3865 (1996).
Desciption of the pseudopotentials used in the r2SCAN related calculations.
All calculations used pseudopotentials from the “PBE PAW datasets version 54” set released in September 2015; a list of the specific POTCAR symbols used for each element is provided below. Although these pseudopotentials were developed for use with the PBE functional, their use with SCAN is common practice because no SCAN-specific pseudopotentials are available for use in VASP.
Element
POTCAR symbol
Ac
Ac
References
[1] R. Kingsbury, A. S. Gupta, C. J. Bartel, J. M. Munro, S. Dwaraknath, M. Horton, and K. A. Persson Phys. Rev. Materials 6, 013801 (2022)
Ag
Ag
Al
Al
Ar
Ar
As
As
Au
Au
B
B
Ba
Ba_sv
Be
Be_sv
Bi
Bi
Br
Br
C
C
Ca
Ca_sv
Cd
Cd
Ce
Ce
Cl
Cl
Co
Co
Cr
Cr_pv
Cs
Cs_sv
Cu
Cu_pv
Dy
Dy_3
Er
Er_3
Eu
Eu
F
F
Fe
Fe_pv
Ga
Ga_d
Gd
Gd
Ge
Ge_d
H
H
He
He
Hf
Hf_pv
Hg
Hg
Ho
Ho_3
I
I
In
In_d
Ir
Ir
K
K_sv
Kr
Kr
La
La
Li
Li_sv
Lu
Lu
Mg
Mg_pv
Mn
Mn_pv
Mo
Mo_pv
N
N
Na
Na_pv
Nb
Nb_pv
Nd
Nd_3
Ne
Ne
Ni
Ni_pv
Np
Np
O
O
Os
Os_pv
P
P
Pa
Pa
Pb
Pb_d
Pd
Pd
Pm
Pm_3
Pr
Pr_3
Pt
Pt
Pu
Pu
Rb
Rb_sv
Re
Re_pv
Rh
Rh_pv
Ru
Ru_pv
S
S
Sb
Sb
Sc
Sc_sv
Se
Se
Si
Si
Sm
Sm_3
Sn
Sn_d
Sr
Sr_sv
Ta
Ta_pv
Tb
Tb_3
Tc
Tc_pv
Te
Te
Th
Th
Ti
Ti_pv
Tl
Tl_d
Tm
Tm_3
U
U
V
V_pv
W
W_sv
Xe
Xe
Y
Y_sv
Yb
Yb_2
Zn
Zn
Zr
Zr_sv
Elastic tensor (original), Cij
GPa
Tensor, describing elastic behavior, unsymmetrized, corresponding to POSCAR (conventional standard cell) orientation
Description of the pseudo-potentials (PSP) used in the GGA and GGA+U calculations.
On 2023-05-02, we changed the Yb PSP in all VASP input sets from Yb_2 to Yb_3 as Yb_2 gives incorrect thermodynamics for most systems with Yb3+. See pymatgen#2968 for details. We are also recomputing all Yb compounds in MP for an upcoming database release. The release notes will highlight this change.
Pseudopotentials are used to reduce computation time by replacing the full electron system in the Coulombic potential by a system only taking explicitly into account the "valence" electrons (i.e., the electrons participating into bonding) but in a pseudopotential. This approach not only reduces the electron number but also the energy cutoff necessary (this is critical in plane-wave-based computations). All computations in the materials project have been performed using a specific type of very efficient pseudopotentials: the projector augmented wave (PAW) pseudopotentials. We used the library of PAW pseudopotentials provided by VASP but for a given element there are often several possibilities in the VASP library. This wiki presents how the choices between the different pseudopotential options were made.
The strategy
As a test set, we ran all elements and binary oxides present in the ICSD with the available PAW pseudopotentials. As it is difficult to test for all properties (structural, electronic, etc...), we chose to be inclusive and to select the pseudopotential with the largest number of electrons (high e) except if convergence issues were seen on our test set, or if previous experience excluded a specific pseudopotential. We also excluded pseudopotentials with too large an energy cutoff.
We also compared to recommendations from the VASP manual present in .
Finally, as we had energies for elements and binary oxides, we compared binary oxide formation energies with the available pseudopotentials. The oxygen molecule energy was obtained from Wang et al. Please note that this data is pure GGA and some chemistries (e.g., transition metals) will give extremely bad formation energy results in GGA. This is not an issue with the pseudopotential but with the functional, so we do not focus on that issue in this wiki.
Pseudopotential comments and choice
1st-row elements
Usually, they have three pseudopotentials: a soft _s, a hard _h, and a standard. The standard is recommended by VASP and will be used for all. The hard ones have extremely high cut-offs (700 eV)
alkali and alkali-earth
The table below indicates our choices. Basically, we chose all high e- pseudopotentials except for Na where we excluded Na_sv due to its very high cutoff (700 eV).
element
options
VASP
Low elec: oxide form_enth (exp-comp) eV per fu
High elec: oxide form_enth (exp-comp) eV per fu
High e- conv. Stats
our choice
rem
d-elements, transition metals
The table below shows the details on the PSP choices. All high e- PSPs have been chosen except for Pd which had convergences problem with the high e- PSP in PdO.
element
options
VASP
Low elec: oxide form_enth (exp-comp) eV per fu
High elec: oxide form_enth (exp-comp) eV per fu
High e- conv. Stats
our choice
rem
main group
Si, P, Cl, S will be used in their standard form (not hard) as suggested by VASP manual.
The Al_h psp was found to be definitely wrong in terms of band structure. There were "ghost" states found in the DOS.
Pb is interesting as the high e- psp shows significantly higher error in formation energies. We kept the high e- psp (Pb_d), but it might be interesting to study this a little more. One hypothesis relies on a recent result showing that lead oxide formation energies need the use of spin-orbit coupling to be accurate. Our computations do not include any relativistic corrections for valence electrons. However, spin-orbit coupling is taken into account during the psp construction. This would explain why a psp with more core electrons (treated indirectly with spin-orbit coupling) would give more accurate results than a psp with fewer electrons.
Bi_d shows a convergence problem, so the decision on Bi has been postponed to further analysis.
Finally, Po and At, while referred to in the VASP manual, are not present in the VASP PAW library.
element
options
VASP
Low elec: oxide form_enth (exp-comp) eV per fu
High elec: oxide form_enth (exp-comp) eV per fu
High e- conv. Stats
our choice
rem
rare-earth, f-electrons
These are probably the most problematic to use as pseudopotentials. Here is what the VASP manual says about them:
Due to self-interaction errors, f-electrons are not handled well by presently available density functionals. In particular, partially filled states are often incorrectly described, leading to large errors for Pr-Eu and Tb-Yb where the error increases in the middle (Gd is handled reasonably well, since 7 electrons occupy the majority shell). These errors are DFT and not VASP related. Particularly problematic is the description of the transition from an itinerant (band-like) behavior observed at the beginning of each period to localized states towards the end of the period. For the elements, this transition occurs already in La and Ce, whereas the transition sets in for Pu and Am for the elements. A routine way to cope with the inabilities of present DFT functionals to describe the localized electrons is to place the electrons in the core. Such potentials are available and described below. Furthermore, PAW potentials in which the states are treated as valence states are available, but these potentials are not expected to work reliable when the electrons are localized.
In summary, the pseudopotentials can either include or not include f electrons; how accurate including them or not is depends on the nature of the bonding for each particular system (localized or not).
What we found is that convergence issues are often seen for high electron psp (e.g., Pr, Nd, Sm). Also, some pseudopotentials (e.g., Er_2, Eu_2) freeze too many electrons and therefore have issues with oxidation states that make one of the frozen electron participate in bonding (e.g., Eu2O3, Er2O3). Finally, there is a major problem with Tb. Only Tb_3 exists but Tb is known to also form Tb4+ compounds (e.g., TbO2). For those Tb4+ compounds, this psp is likely to be extremely wrong. There is currently no fix for this except waiting for someone to develop a PAW Tb_4 psp.
element
options
VASP
Low elec: oxide form_enth (exp-comp) eV per fu
High elec: oxide form_enth (exp-comp) eV per fu
High e- conv. Stats
our choice
rem
transuranides, f-electrons
U, Ac, Th, Pa, Np, Pu, Am
Following VASP suggestion, we decided to use the standard (and not the soft) version for all those pseudopotentials.
Citation
To cite the Materials Project, please reference the following work:
A. Jain, G. Hautier, C. J. Moore, S. P. Ong, C. C. Fischer, T. Mueller, K. A. Persson, and G. Ceder, A high-throughput infrastructure for density functional theory calculations, Computational Materials Science, vol. 50, 2011, pp. 2295-2310.
Authors
Geoffroy Hautier
References
[1]: P.E. Blöchl, Physical Review B 50, 17953-17979 (1994).
[2]: R. Ahuja, A. Blomqvist, P. Larsson, P. Pyykkö, and P. Zaleski-Ejgierd, Physical Review Letters 106, 1-4 (2011).
0.03
0.01
all converged
Li_sv
highest e- psp chosen
Na
Na, Na_sv, Na_pv
Na_pv
0.06
0.01
all converged
Na_pv
Na_sv is extremely high in cutoff (700 eV) for marginal gain in accuracy on Na2O
K
K_pv, K_sv
K_sv
0.01
0.01
80% conv for both
K_sv
highest e- psp chosen
Cs
Cs_sv
Cs_sv
Cs_sv
Rb
Rb_pv, Rb_sv
Rb_sv
0.05
0.03
all converged
Rb_sv
highest e- psp chosen
Be
Be, Be_sv
Be
0.04
0.04
all converged
Be_sv
highest e- psp chosen
Mg
Mg, Mg_pv
Mg_pv
0.02
0.05
all converged
Mg_pv
VASP and thermo suggest Mg as they are not much different; we decided to stick with the high e- psp.
Ca
Ca_sv, Ca_pv
Ca_pv
0.06
0.03
all converged
Ca_sv
highest e- psp chosen
Sr
Sr_sv
Sr_sv
Sr_sv
Ba
Ba_sv
Ba_sv
Ba_sv
Sc_sv
Y
Y_sv
Y_sv
Y_sv
Ti
Ti, Ti_pv, Ti_sv
Ti_pv
0.13
0.23
metal conv pb with Ti and Ti_sv
Ti_pv
highest e- psp with best conv. chosen
Zr
Zr, Zr_sv
Zr_sv
0.06
0.03
all converged
Zr_sv
highest e- psp chosen
Hf
Hf, Hf_pv
Hf_pv
0.19
0.18
all converged
Hf_pv
highest e- psp chosen
V
V, V_pv, V_sv
V_pv
0.39
0.46
all converged
V_sv
highest e- psp chosen
Nb
Nb_pv
Nb_pv
Nb_pv
Ta
Ta, Ta_pv
Ta_pv
0.3
0.31
similar conv. for both
Ta_pv
highest e- psp chosen
Cr
Cr, Cr_pv
Cr_pv
0.53
0.6
all converged
Cr_pv
highest e- psp chosen
Mo
Mo, Mo_pv
Mo_pv
0.39
0.45
all converged
Mo_pv
highest e- psp chosen
W
W, W_pv
W_pv
0.47
0.48
all converged
W_pv
highest e- psp chosen
Mn
Mn, Mn_pv
Mn or Mn_pv (!)
0.29
0.31
all converged
Mn_pv
highest e- psp chosen
Tc
Tc, Tc_pv
Tc or Tc_pv
all converged (no metals BTW)
Tc_pv
highest e- psp chosen
Re
Re, Re_pv
Re
0.56
0.59
all converged
Re_pv
highest e- psp chosen
Fe
Fe, Fe_pv
Fe_pv
0.62
0.47
50% conv. on oxides for both psp
Fe_pv
highest e- psp chosen
Co
Co
Co
Co
Ni
Ni, Ni_pv
Ni
0.4
0.4
all converged
Ni_pv
highest e- psp chosen
Cu
Cu, Cu_pv
Cu
0.07
0.1
all converged
Cu_pv
highest e- psp chosen
Zn
Zn
Zn
Zn
Ru
Ru, Ru_pv
Ru
0.41
0.41
all converged
Ru_pv
highest e- psp chosen
Rh
Rh, Rh_pv
Rh
0.36
0.35
all converged
Rh_pv
highest e- psp chosen
Pd
Pd, Pd_pv
Pd
0.2
0.2
Pd_pv has one unconv. PdO
Pd
due to the conv. issue we chose Pd (recommended by VASP too).
Ag
Ag
Ag
Cd
Cd
Cd
Hg
Hg
Hg
Au
Au
Au
Ir
Ir
Ir
Pt
Pt
Pt
Pt
Os
Os, Os_pv
Os_pv
0.67
0.7
all converged
Os_pv
highest e- psp chosen
0.05
0.01
all converged
Ga_d
Ga_h seems best (0.01 instead of 0.02) but same problem as Al_h?
Ge
Ge, Ge_d, Ge_h
Ge_d
0.06
0.06
all converged
Ge_d
Ge_h seems best (Ge_h and Ge_d similar though) but same problem as Al_h ?
Al
Al, Al_h
Al
0.03
0.01
all converged
Al
Good energetics but pb in band structure
As
As
Se
Se
Br
Br
In
In, In_d
In_d
0.13
0.1
all converged
In_d
highest e- psp chosen
Sn
S, Sn_d
Sn_d
0.16
0.12
all converged
Sn_d
highest e- psp chosen
Tl
Tl, Tl_d
Tl_d
0.26
0.31
all converged
Tl_d
highest e- psp chosen
Pb
Pb, Pb_d
Pb_d
0.17
0.36
all converged
Pb_d
highest e- psp chosen
Bi
Bi, Bi_d
Bi_d
convergence pb
?
Po
Po, Po_d
Po
no Po psp is available in the PAW library!
At
At, At_d
At_d
no At psp is available in the PAW library
0.12
0.17
all converged
La
La_s means soft
Ce
Ce_3, Ce
/
1.18
0.26
all converged
Ce
thermo data on CeO2 is terrible with Ce_3, cf Ce4+ thermo data on Ce2O3 is similar with both
Pr
Pr_3, Pr
/
0.00
0.09
Pr metal did not converge
Pr_3
Pr_3 better oxide thermo (surprisingly good!) and convergence in metal.
Nd
Nd_3, Nd
/
0.04
0.01
Nd metal conv. problem
Nd_3
convergence pb
Pm
Pm_3, Pm
/
/
/
Pm_3
no real data to compare, it is between Nd and Sm in the periodic table, so we decided to pick a _3 as Nd and Sm
Sm
Sm_3, Sm
/
0.1
/
Sm metal conv. pb
Sm_3
conv pb
Eu
Eu_2, Eu
/
0.68
0.25
all converged
Eu
Both EuO and Eu2O3 thermo worse with Eu_2
Gd
Gd_3, Gd
/
0.2
0.12
all converged
Gd
Gd has better thermo and highest e-
Tb
Tb_3
/
all converged
Tb_3
There is a major pb with Tb. It can 4+ and we have only a 3+ psps
Dy
Dy_3
/
all converged
Dy_3
Ho
Ho_3
/
Ho_3
Er
Er_2, Er_3
/
1.16
0.15
all converged
Er_3
thermo data on Er2O3 off with Er_2
Tm
Tm, Tm_3
/
0.2
?
could not converge any metal with Tm
Tm_3
Yb
Yb_3, Yb_2, Yb
/
1.03
0.59
all converged
Yb_3
thermo data off with Yb_2 and Yb has convergence issues